如何利用基因编辑技术研究基因点突变
如何利用基因编辑技术研究基因点突变?

在精准医学快速发展的今天,基因点突变的研究成为揭示人类疾病机制和开发靶向疗法的关键方向。借助CRISPR/Cas9等前沿基因编辑技术,科学家可以在细胞或动物模型中精确引入单个核苷酸变化,从而模拟疾病发生的分子机制,为基础研究和转化医学提供了有力工具。
什么是点突变?
点突变指DNA序列中单个碱基的改变(包括替换、插入或缺失一个碱基)。这类突变可能发生在蛋白编码区(外显子)或非编码区(如启动子、增强子、内含子等调控区域),并按功能影响可分为功能丧失型与功能获得型。如图所示,编码区点突变可分为同义突变(不改变氨基酸)和非同义突变(改变氨基酸,如错义或无义),非编码区点突变虽不直接改变蛋白序列,但可能影响基因表达或RNA剪接调控。功能丧失型突变使基因产物失去正常功能,多见于隐性遗传病;功能获得型突变则赋予基因新活性或增高活性,常与显性疾病或肿瘤相关。
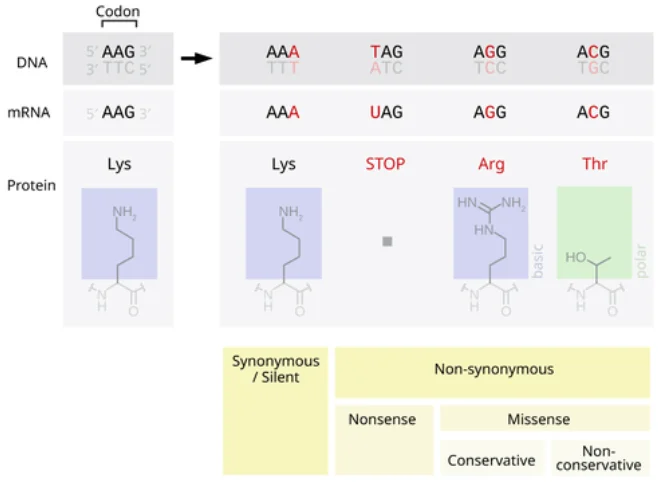
图1. 点突变对蛋白质影响的分类示意图。左侧同义突变不改变氨基酸(Lys→Lys),右侧包括无义突变(产生终止密码子Tag,截短蛋白)和错义突变(AA→Arg/Thr,改变氨基酸)。红字为突变碱基,蓝色和绿色框为氨基酸结构示意。
1. 编码区点突变:位于基因外显子,分为同义突变(蛋白质序列不变)和非同义突变(改变氨基酸,引发结构或功能变化)。
2. 非编码区点突变:位于启动子、增强子、剪接位点或UTR等区域,不改变蛋白序列,但可能影响基因表达水平、RNA剪接或调控元件功能。
3. 功能丧失突变:基因产物失活或截短,如产生无义密码子导致蛋白功能丧失。此类突变多为隐性遗传(需双等位基因突变才能致病)。
4. 功能获得突变:突变使基因获得新功能或过度活化,常见于显性疾病或癌症基因,如原癌基因突变让细胞持续增殖。
CRISPR/Cas9 构建点突变模型的原理
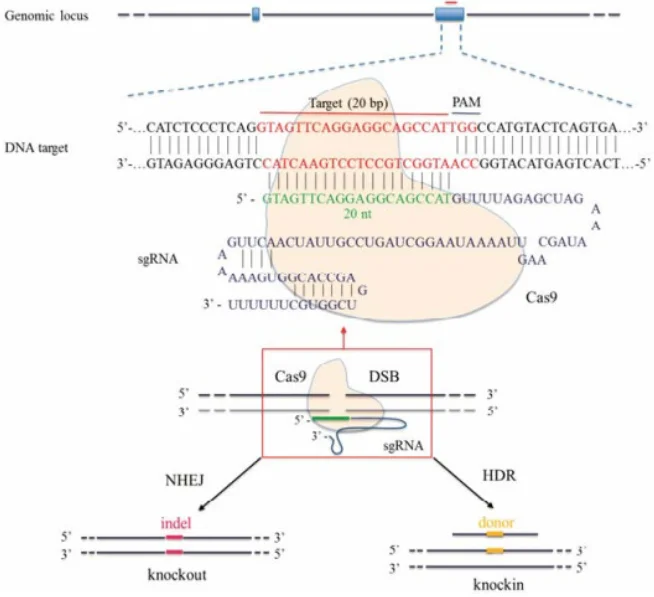
图2:CRISPR/Cas9介导的基因编辑示意图。Cas9蛋白(黄色)在sgRNA引导下识别蓝色的靶序列并切割DNA造成双链断裂(DSB)。细胞启动修复机制,非同源末端连接(NHEJ)常引入小插入/缺失(indel)导致基因敲除(左),而同源重组(HDR)则利用提供的供体DNA(黄色片段)精确修复并插入特定突变(右)
CRISPR/Cas9系统能够在基因组特定位点造成双链断裂(DSB),并借助细胞自身修复机制实现基因组改造。如上图所示,Cas9在单向导RNA(sgRNA)靶向下切割DNA后,若不提供供体模板,细胞通过NHEJ修复往往产生随机的插入/缺失突变(indel),可导致编码框移位或提前终止,实质上敲除了该基因。若实验中同时引入含有期望突变的同源重组修复模板(如含突变位点的双链DNA或单链寡核苷酸),细胞可通过HDR精确地将该突变“修复”到基因组中,从而实现点突变。
利用这一原理,我们可以构建携带特定点突变的细胞系或动物模型:设计一对sgRNA/Cas9靶向目标位点,并提供包含目标突变的供体DNA后,细胞的HDR修复机制会将突变导入基因组。获得编辑后的混合细胞后,通常通过单克隆筛选等方式分离得到携带纯合或杂合突变的克隆株。在多倍体细胞或动物胚胎中,若编辑效率高,可能同时编辑两个等位基因,获得纯合突变;若只编辑一个等位基因,则得到杂合突变模型。对于显性遗传疾病,往往只需要构建杂合突变即可模拟患者基因型;对于隐性遗传疾病,则需要筛选出含双等位基因突变的纯合模型。
疾病模型案例
中心核肌病(CNM,杂合突变):
常染色体显性CNM常由DNM2基因的单点突变(如R465W)引起。Rabbi等人在患者来源的成肌母细胞和小鼠模型中设计了只识别突变等位基因的sgRNA/Cas9体系,成功靶向并敲除了突变的DNM2等位基因。编辑后,原本异常的细胞膜转运和自噬等功能恢复正常。这说明CRISPR/Cas9可以对显性突变等位基因进行等位特异性编辑,从而逆转疾病细胞的表型,为显性点突变疾病的治疗和研究提供了可能。
阿尔茨海默病(AD,杂合突变):
家族性早发AD常由APP或PSEN基因的点突变引发,如瑞典突变(APP KM670/671NL)或PSEN1 M146L突变。研究人员利用CRISPR/Cas9设计了特异性gRNA,只破坏突变等位基因。在携带瑞典突变的患者成纤维细胞中,敲除突变的APP等位基因使培养上清中Aβ肽的水平降低约。类似地,在携带PSEN1 M146L异源突变的细胞中进行基因编辑后,原本升高的Aβ42/40比例显著下降并部分恢复正常。这些结果表明,针对AD相关点突变的CRISPR编辑可减轻致病性表型,为阿尔茨海默病机制研究和治疗探索提供新思路。
Tay-Sachs病(TSD,纯合突变):
TSD是一种常染色体隐性遗传的神经变性病,80%的患者携带HEXA基因第11外显子四碱基(TATC)插入突变nature.com。在小鼠中敲除HEXA基因并未能重现典型病理,为此Qian等人尝试使用基因编辑技术构建TSD模型。他们最终采用了新一代引物编辑技术在兔子HEXA基因中插入TATC突变,成功获得了携带该插入突变的兔模型,并观察到了肌无力、共济失调等与人类TSD相似的表型。这一实例说明,通过精准编辑将人类致病点突变导入动物模型中,可在体内重现相应遗传病表型,为深入研究疾病机制提供了工具。
囊性纤维化(CF,纯合突变):
CF是因CFTR基因隐性突变导致的致死性遗传病,ΔF508等点突变会使CFTR蛋白功能丧失。Fan等人在绵羊中应用CRISPR/Cas9和体细胞核移植技术,敲除了绵羊CFTR基因。他们成功获得了CFTR^–/–和CFTR^+/–的绵羊,出生后CFTR^–/–绵羊出现胰腺纤维化、肠梗阻、输精管缺失等与人类CF高度一致的病理表现。这一大型动物模型展示了CFTR基因缺失足以诱发典型CF表型,验证了该模型用于测试新药和基因疗法的潜力。
点突变疾病模型的意义
利用CRISPR/Cas9技术构建携带特定点突变的疾病模型,有助于精准模拟人类病变基因型,为研究疾病发病机制和验证治疗方案提供了强大平台。正如国内学者指出,CRISPR/Cas9实现了对靶基因的定点编辑,使得构建动物或细胞系模型更加便捷,大大加速了药物靶点筛选、验证和新药研发。例如上述CF绵羊模型和TSD兔模型不仅重现了人类疾病的关键病理,还可用于体内评价针对突变的基因治疗或药物疗效。现代新药研发越来越依赖精准疾病模型:通过点突变模型,我们可以在发病机制研究阶段识别关键信号通路,在药物筛选阶段测试候选药物对特定突变的疗效,从而为个性化医疗和基因治疗策略的开发奠定基础。
新型基因编辑平台的未来潜力
随着技术不断迭代,新一代基因编辑工具正为点突变研究带来更多可能。源井生物推出的CRISPR-U™平台和EZ-HRex™,实现基因编辑效率比传统CRISPR/Cas9高出10~20倍,可高效获得基因敲除、点突变、基因敲入。此外,碱基编辑(Base Editing)和引物编辑(Prime Editing)等技术能够在不造成DNA双链断裂的情况下直接实现单碱基替换,为修正或模拟点突变提供了新途径。未来,这些高效、精准的基因编辑平台将极大简化疾病模型的构建过程,使研究人员能更快速地生成各种复杂突变背景下的细胞系和动物模型。这不仅加速了基础科研,也为基因治疗和个体化药物研发拓展了新途径。

图3:源井EZ-HRex™新技术
综上所述,基因编辑技术特别是CRISPR/Cas9及其衍生平台,为点突变的研究提供了强有力的工具。从突变机制探索到模型构建和药物筛选,科学家们正充分利用这些技术推进生命科学研究,并展望在不久的将来,通过这些新兴平台可以更高效地破解遗传性疾病,为精准医学和创新疗法的发展创造更多可能。
参考文献
[1] Rabai A, Reisser L, Reina-San-Martin B, Mamchaoui K, Cowling BS, Nicot AS, Laporte J. Allele-Specific CRISPR/Cas9 Correction of a Heterozygous DNM2 Mutation Rescues Centronuclear Myopathy Cell Phenotypes. Mol Ther Nucleic Acids. 2019 Jun 7;16:246-256. doi: 10.1016/j.omtn.2019.02.019. Epub 2019 Feb 27. PMID: 30925452; PMCID: PMC6439232.
[2] Konstantinidis E, Molisak A, Perrin F, Streubel-Gallasch L, Fayad S, Kim DY, Petri K, Aryee MJ, Aguilar X, György B, Giedraitis V, Joung JK, Pattanayak V, Essand M, Erlandsson A, Berezovska O, Ingelsson M. CRISPR-Cas9 treatment partially restores amyloid-β 42/40 in human fibroblasts with the Alzheimer's disease PSEN1 M146L mutation. Mol Ther Nucleic Acids. 2022 Mar 28;28:450-461. doi: 10.1016/j.omtn.2022.03.022. PMID: 35505961; PMCID: PMC9043867.
[3] György B, Lööv C, Zaborowski MP, Takeda S, Kleinstiver BP, Commins C, Kastanenka K, Mu D, Volak A, Giedraitis V, Lannfelt L, Maguire CA, Joung JK, Hyman BT, Breakefield XO, Ingelsson M. CRISPR/Cas9 Mediated Disruption of the Swedish APP Allele as a Therapeutic Approach for Early-Onset Alzheimer's Disease. Mol Ther Nucleic Acids. 2018 Jun 1;11:429-440. doi: 10.1016/j.omtn.2018.03.007. Epub 2018 Mar 16. PMID: 29858078; PMCID: PMC5992788.
[4] 4.Fan, Z., et al. "A sheep model of cystic fibrosis generated by CRISPR/Cas9 disruption of the CFTR gene. JCI Insight 3: e123529." 2018






