基因敲除细胞系构建指南:如何高效设计gRNA实现彻底KO,避免蛋白残留

基因敲除细胞系构建指南:如何高效设计gRNA实现彻底KO,避免蛋白残留

基因组编辑技术,特别是以CRISPR/Cas9为代表的方法,正在彻底改变基因功能研究的路径。作为当前最主流的基因编辑工具,CRISPR/Cas9系统凭借其高效率、灵活性和操作便捷性,在基因敲除(Knockout, KO)领域发挥了关键作用,显著推动了基础科研与应用研究的进展。
然而,在构建基因敲除细胞系的过程中,如何设计高效gRNA并有效避免蛋白残留,仍是影响KO成功率的核心技术瓶颈。本文将系统剖析这些关键挑战,并分享源井生物在该领域积累的实用经验与技术解决方案。
CRISPR/Cas9介导基因敲除的基本原理
在CRISPR/Cas9系统中,Cas9核酸酶在向导RNA(gRNA)的引导下精准切割DNA,形成双链断裂(DSB)。随后,细胞通过非同源末端连接(NHEJ)修复断裂,但这一修复方式常导致碱基插入或缺失(Indels),从而引发移码突变,提前出现终止密码子,实现基因功能敲除。然而,在实际应用中,即便在DNA水平成功敲除目标序列,在蛋白质水平仍可能出现异常表达。主要原因包括:(1)翻译重新启动(Reinitiation),(2)外显子跳跃(Exon Skipping)。
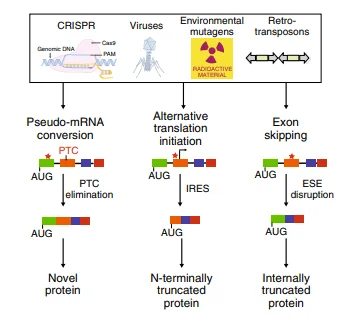
图1: 抗INDEL效应导致CRISPR基因敲除实验失败的细胞机制
隐患一:翻译重新启动(Translation Reinitiation)导致的蛋白截短表达
尽管敲除位点移码,但有研究发现,当突变发生在ORF 5′端附近时,mRNA降解不完全,翻译可能在下游的起始密码子重新启动。也就是说,核糖体在遇到提前终止后并未完全解离,而在同一mRNA的内部AUG重新起始翻译,产生N端截短蛋白。这种“非法翻译(Illegitimate Translation, ITL)”现象已在多个案例中观察到,例如在针对5′端的Frameshift突变中,后续下游AUG被启动产生带N端缺失的蛋白,仍保留一定功能。因此,单纯依靠近起始密码子的靶点并不总能避免残留蛋白。如果内部AUG启动位点所在的内含子或外显子未被破坏,就可能产生可被抗体识别的截短蛋白,导致后续Western Blot验证出现阳性条带。已有综述指出,翻译重新启动和可变剪接是CRISPR敲除逃逸产生残留蛋白的主要机制。
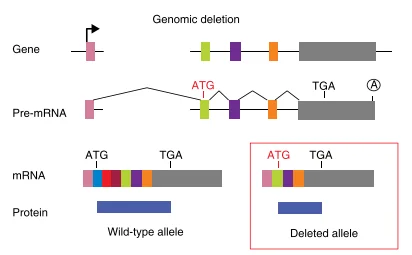
图2: 翻译重新启动的机制
隐患二:外显子跳跃导致的异常剪接
CRISPR引入indel后还可能影响前体mRNA剪接,从而跳过被编辑的外显子。经典研究表明,单个gRNA靶向外显子时,会通过两种独立机制导致外显子跳跃:其一是在突变的pre-mRNA剪接过程中直接跳过该外显子;其二是由于CRISPR可能诱导大片段缺失(删除多个外显子),剩余外显子重新组合剪接。示例图见图三:CRISPR诱导的大片段缺失将前几个外显子完全去除,使后续Exon上的内部AUG成为新的起始位点,从而翻译出N端截短蛋白。此外,即使只有单个外显子的少量插缺突变,也能破坏外显子的剪接增强子(ESE)或引入剪接沉默子(ESS),促使该外显子在剪接时被跳过。值得注意的是,如果目标外显子长度为3的倍数(对称外显子),其跳过后不会移码,反而有可能产生功能性缺失蛋白。综上,外显子跳跃可恢复阅读框并表达内部缺失蛋白,从而干扰敲除验证和后续功能分析。
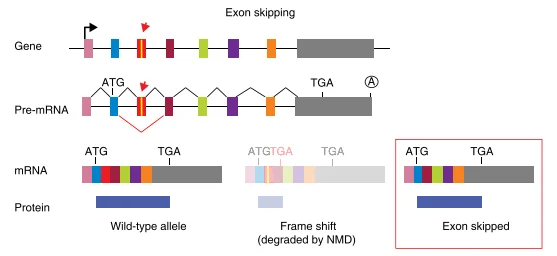
图3: 外显子跳跃的机制
如何制定科学有效的基因敲除策略?
为了最大程度避免上述问题,需要在设计gRNA时综合考虑多个因素:
1. 效率预测:应借助成熟工具评估gRNA靶向效率。例如,当前已有多种计算工具通过机器学习模型预测gRNA切割效率,可排除结合差或二级结构阻碍的gRNA。一般优选GC含量适中、无大规模二级结构且靶序列独特的gRNA以提高敲除效率。
2. 脱靶风险评估:使用离线/在线算法检测gRNA在基因组中的相似位点,避免选择在其他重要基因中有高同源性的序列。通过比对全基因组序列,可对候选gRNA的潜在脱靶位点进行打分筛选。
3. 外显子特性分析:分析目标基因的转录本和外显子结构,确定关键外显子。在潜在存在内部起始密码子或多个翻译框的基因中,应优先靶向覆盖所有可能ORF的大区域;如果存在对称外显子,应避免仅靶向这些外显子(因跳过后会保留功能)。同时需参考已有文献和数据库,明确关键功能域所在外显子,确保敲除后该功能域被破坏。
4. 多gRNA策略:若单个gRNA难以实现全面敲除,可同时设计两个或多个gRNA。例如,使用两条gRNA切割同一基因的不同比邻位点,诱导大片段缺失,从而保证移码,或覆盖多个外显子增加敲除成功率。多sgRNA策略也可用于构建不同突变组合,以排除单点gRNA可能的遗漏。
在gRNA设计阶段,需要平衡靶向效率和安全性,力求找到最佳靶点组合,减少后续“意外惊喜”的概率。例如,如果存在内含AUG启动的内部ORF,应确保至少一个gRNA能够破坏该内部ORF;如果担心外显子跳跃,应优先靶向多个不同外显子或采用跨外显子的双gRNA。总之,仔细分析靶基因的转录本和剪接结构,是设计高质量敲除策略的关键。
KO验证策略
构建敲除细胞系后,需要多层次验证来确认敲除效果
1. DNA水平验证:首先通过PCR扩增靶基因所在区域并进行测序(Sanger或深度测序分析)来确认indel的存在及序列改变。T7E1酶切检测或TIDE分析也可快速评估突变频率。只有目标序列发生预期插缺或大片段删除时,才可认定基因被成功破坏。
2. RNA水平验证:采用RT-qPCR或RNA测序检测目标基因的mRNA表达情况。若mRNA显著下调且不出现新的剪接变体,表明突变转录本被NMD清除。否则,应设计引物跨接突变外显子上下游,通过RT-PCR检测是否存在异常剪接产物(如缺失目标外显子的mRNA)。如果发现跳跃外显子或内源启动转录产物,应结合序列比对确认其来源。
3. 蛋白水平验证:最终通过蛋白质检测确认敲除。常用Western blot检测目的蛋白表达,预期应仅见野生型分子量条带消失。如仍见预期位置以外的信号条带,提示可能存在截短或融合蛋白。此时可结合质谱鉴定这些未知条带的肽段序列,确定其来源ORF。总之,多级验证(基因组、转录和蛋白)缺一不可,以确保获得真正无表达残留的敲除细胞株。
源井生物专业解决方案
针对上述挑战,源井生物凭借丰富经验和创新技术为科研工作者提供可靠的敲除细胞系构建服务。公司15年专注基因编辑技术研发和服务,率先推出自主研发的红棉CRISPR基因编辑系统,集合了千例大数据和自动化方案设计能力。我们已在500多种细胞系上累计完成10000余例CRISPR实验,服务领域遍及全球科研机构。特别是在基因敲除项目中,源井团队已帮助客户在500余种细胞系构建敲除系,集成了从gRNA设计、质粒/病毒载体构建到单克隆挑选和多层次验证的全流程方案。依托历年累积的数据分析,我们为科研人员提供5000多例已通过Western等验证的敲除细胞系案例参考,并可定制多gRNA或大片段删除方案,最大程度避免蛋白残留问题。
综上所述,成功构建高质量的基因敲除细胞系,不仅依赖于高效的敲除策略,更需要对潜在的脱靶效应及基因残留活性进行严谨评估。通过精准的gRNA设计、多层面的验证手段,并借助源井生物成熟的“红棉”优化系统与专业服务平台,研究人员能够有效规避蛋白残留等风险,从而高效、可靠地获得理想的基因完全敲除细胞模型,为下游功能研究奠定坚实基础。
参考文献
[1] Biological plasticity rescues target activity in CRISPR knockouts. Nature methods (2019), 16(11):1087-1093.
[2] Low Incidence of Off-Target Mutations in Individual CRISPR-Cas9 and TALEN Targeted Human Stem Cell Clones Detected by Whole-Genome Sequencing. Cell Stem Cell (2014),15:27–30.
[3] Agreement between two large pan-cancer CRISPR-Cas9 gene dependency data sets. Nat Commun (2019), 10(1):5817.
[4] Unexpected consequences: exon skipping caused by CRISPR-generated mutations. Genome Biology (2017) 18:109.
[5] Design and analysis of CRISPR–Cas experiments.Nature biotechnol (2020) 38(7):813-823.







