突破耐药壁垒!CRISPR 转录因子文库筛出 SMAD1,直击 CRC 潘氏样转化根源!

引言
结直肠癌(CRC)中 KRAS 突变率高达 50%,KRAS 与 EGFR 双靶向治疗虽展现出 34%-46% 的客观缓解率,但耐药问题仍严重制约疗效,多数患者数月内复发。2025 年 11 月,中山大学肿瘤防治中心高益军团队等人在 Cancer Cell 发表最新研究。该研究整合多类模型与临床样本,通过 CRISPR 转录因子文库筛选锁定核心调控因子 SMAD1, 首次发现 CRC 细胞在治疗压力下会发生类潘氏转化以逃避免疫。进一步证实 SMAD1-FGFR3 信号轴是驱动该谱系可塑性及 MAPK 通路再激活的关键,联合抑制 FGFR3 可阻断类潘氏转化、恢复治疗敏感性。CRISPR 筛选为耐药机制破解提供精准靶点,该发现为克服 KRAS 突变 CRC 耐药提供了全新靶向策略,临床转化价值显著。

研究背景
KRAS 突变结直肠癌长期面临治疗手段有限、预后较差的困境,近年来 KRAS 抑制剂(如 G12C 抑制剂索托拉西布、G12D 抑制剂 MRTX1133)与 EGFR 抗体的联合疗法成为突破,但耐药机制尚未完全阐明。已知耐药机制包括 KRAS 二次突变、上下游基因改变等基因组层面变化,但约三分之一复发肿瘤无明显基因组异常,提示非遗传机制(如谱系可塑性)可能发挥关键作用。肠道细胞具有高度可塑性,CRC 细胞也可通过状态重编程逃避治疗压力,但 KRAS/EGFR 双靶向治疗诱导 CRC 细胞发生谱系转化的具体形式及分子机制,目前仍不明确,亟待深入探究。
研究目的
明确 KRAS/EGFR 双靶向治疗下 CRC 细胞是否通过谱系可塑性产生耐药,揭示其具体细胞状态变化及核心调控机制,验证靶向该机制能否逆转耐药,为 KRAS 突变 CRC 提供新的治疗方案。
研究方法
- 实验模型: 使用 iKAP KRAS G12D 突变结直肠癌基因工程小鼠模型、KRAS G12D/G12C 突变 CRC 细胞系(LS174T、SW1463 等)、患者来源类器官(PDO)及异种移植模型(PDX),结合临床患者配对活检样本。
- 谱系追踪: 构建 DEFA5 启动子驱动的 Cre-LoxP 重组系统(dsRed-STOP-eGFP)和 CRISPR-Cas9 介导的 DEFA5-CopGFP 敲入系统,追踪类潘氏细胞的起源与动态变化。
- CRISPR筛选: 针对 1741 个人类转录因子进行 CRISPR 敲除筛选,鉴定驱动类潘氏转化的关键调控因子。
- 分子机制验证: 通过 RNA-seq、单细胞 RNA-seq(scRNA-seq)分析基因表达变化;利用 ChIP-qPCR 验证 SMAD1 与 FGFR3 启动子的结合;采用 Western blot、免疫荧光检测蛋白表达及定位;通过报告基因实验分析 DNA 修复通路活性。
- 药物干预: 使用 KRAS 抑制剂(MRTX1133、索托拉西布)、EGFR 抗体(西妥昔单抗)、FGFR 抑制剂(futibatinib、infigratinib),在细胞、类器官及动物模型中验证单药或联合治疗效果。
- 临床样本验证: 分析 2 例接受 KRAS/EGFR 双靶向治疗的 CRC 患者治疗前后的活检样本,检测类潘氏细胞标志物表达。
研究路线
- 耐药表型发现: 在 GEMM、PDO 及 PDX 模型中,观察 KRAS/EGFR 双靶向治疗后残留肿瘤的细胞状态变化,鉴定类潘氏细胞富集现象。
- 谱系可塑性验证: 通过谱系追踪实验,明确类潘氏细胞来源于 CRC 细胞的转分化,而非预存类潘氏细胞的扩增。
- 关键调控因子筛选: 利用 CRISPR 转录因子文库筛选,结合公共队列数据分析,锁定 SMAD1 为类潘氏转化的核心驱动因子。
- 分子机制解析: 验证 SMAD1 对类潘氏转化及耐药的调控作用; 鉴定 SMAD1 下游靶标 FGFR3,明确 SMAD1-FGFR3 信号轴的激活机制; 证实 FGFR3 通过促进 MAPK 通路再激活介导耐药。
- 治疗策略验证: 在细胞、类器官及动物模型中,验证 FGFR 抑制剂与 KRAS/EGFR 双靶向治疗的联合疗效。
- 临床相关性验证: 通过患者配对样本,确认类潘氏转化在临床耐药肿瘤中的富集。
主要结果
-
KRAS/EGFR 双靶向治疗诱导 CRC 细胞发生类潘氏转化: 转录组分析显示,iKAP 小鼠模型经 MRTX1133 + 西妥昔单抗治疗后,残留肿瘤中防御素、抗菌肽等潘氏细胞特征通路显著富集。GSEA 分析证实,潘氏细胞特征基因是治疗后最显著富集的上皮细胞谱系特征(NES=2.4,调整 p<0.001),DEFA5、DEFA6 等标志物表达显著上调。免疫荧光显示,治疗后 iKAP 肿瘤、LS174T 3D 球体及 KRAS G12D/G12C PDO 中,DEFA5 + 类潘氏细胞比例明显增加。scRNA-seq 揭示,CRC 细胞经治疗后先过渡到滞育样药物耐受状态(DTP),再逐步分化为类潘氏细胞(C5 集群),该集群随治疗时间逐渐扩张。
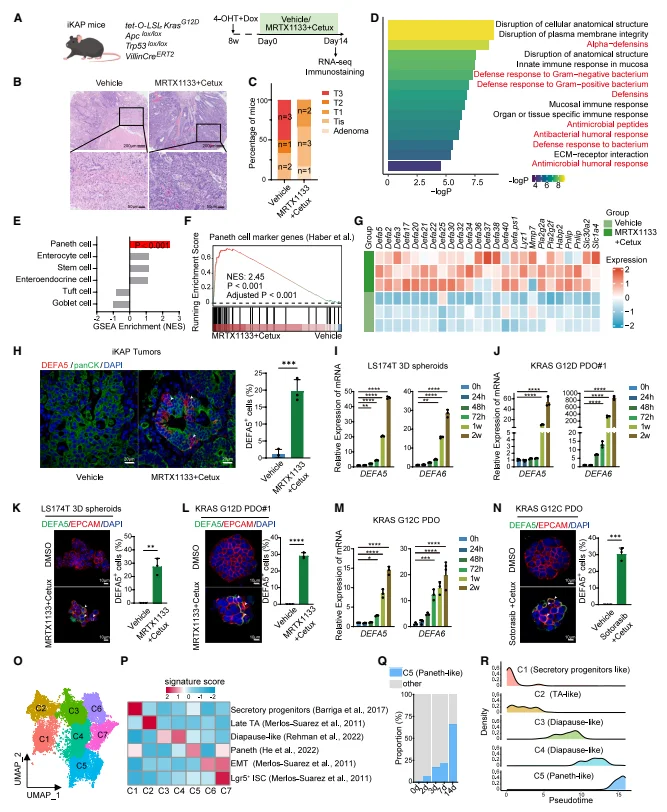
图1. KRAS- EGFR 抑制后残余结直肠癌(CRC)病灶中类潘氏细胞状态的富集
-
类潘氏转化通过转分化介导治疗耐药 谱系追踪显示,治疗前仅 0.23% 的 LS174T 细胞表达 eGFP(类潘氏细胞标志物),治疗后 21 天该比例升至 32.26%,证实类潘氏细胞来源于 CRC 细胞转分化而非预存细胞扩增。药物撤去后,类潘氏细胞比例快速下降,表明该状态依赖治疗压力维持;且分选后的 eGFP + 类潘氏细胞对双靶向治疗的敏感性显著低于 eGFP - 细胞。构建 DEFA5 启动子驱动的 iCasp9 系统,选择性消融类潘氏细胞后,肿瘤对 MRTX1133 + 西妥昔单抗的敏感性显著提升,证实类潘氏细胞是耐药核心群体。类潘氏细胞中 KRAS-GTP 水平及 pERK 表达更高,MAPK 通路再激活是其耐药的关键原因。
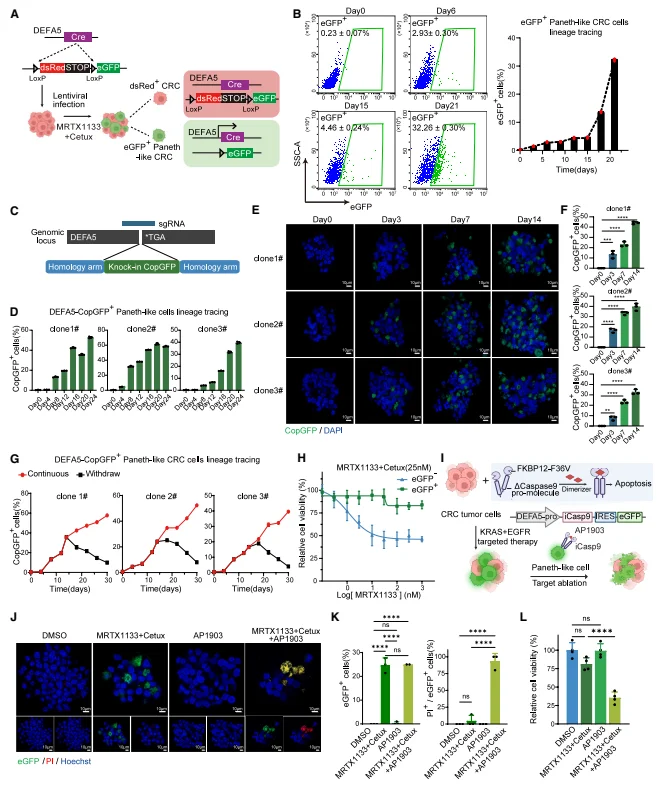
图2. 通过跨分化响应KRAS与 EGFR 联合抑制产生类潘氏细胞
-
CRISPR 筛选鉴定 SMAD1 为类潘氏转化的关键驱动因子 针对 1741 个转录因子的 CRISPR 敲除筛选,发现 34 个基因敲除后可增强肿瘤对双靶向治疗的敏感性,其中 SMAD1 与潘氏细胞特征在 CRC 公共队列中呈显著正相关(r=0.66,p=9.1e-05)。双靶向治疗后,LS174T 球体、KRAS G12D/G12C PDO 中 SMAD1 转录水平显著上调,且 SMAD1 与类潘氏细胞标志物 DEFA5 存在共定位。
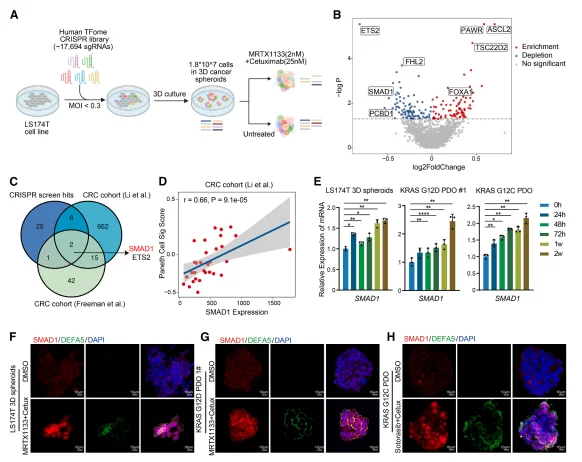
图3.CRISPR 筛选显示SMAD1是类潘氏细胞状态转换及治疗耐药的驱动因子
-
SMAD1 通过驱动类潘氏转化介导耐药 SMAD1 敲除显著增强 LS174T、LS180 细胞对双靶向治疗的敏感性,克隆形成能力下降,体内异种移植模型中 70%-90% 的肿瘤实现退缩。竞争生长实验显示,SMAD1 敲除的 EGFP + 细胞在治疗压力下比例显著降低,表明 SMAD1 缺失使细胞无法耐受双靶向治疗。 SMAD1 敲除显著抑制治疗诱导的 DEFA5、DEFA6 表达及类潘氏细胞形成,而 SMAD1 过表达则上调潘氏细胞标志物并增强耐药性。机制上,SMAD1 的作用不依赖于经典 BMP-SMAD4 通路,SMAD4 敲除不影响类潘氏转化及治疗敏感性。
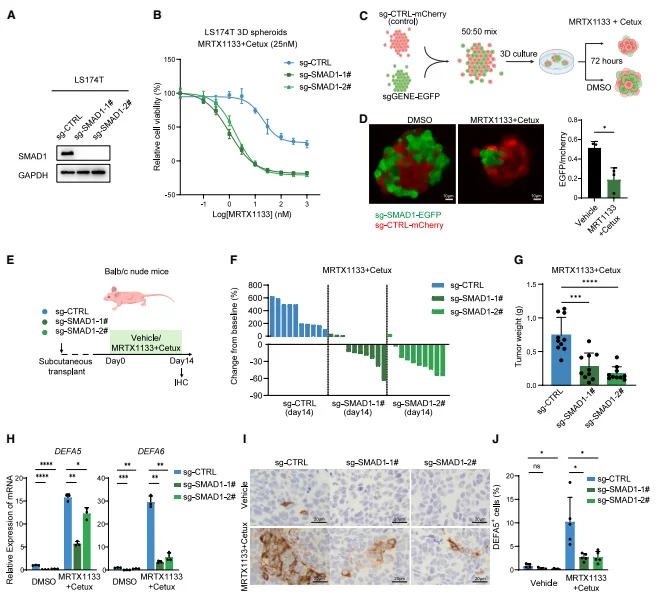
图4. SMAD1通过促进类潘氏状态转换驱动对KRAS- EGFR 联合治疗的耐药性
-
SMAD1-FGFR3 信号轴驱动类潘氏转化及 MAPK 再激活 通路富集分析显示,SMAD1 相关基因集中于 FGFR 信号通路,双靶向治疗后 FGFR3(而非 FGFR1)转录及磷酸化水平显著上调,且与 SMAD1 表达正相关。ChIP-qPCR 证实,SMAD1 可直接结合 FGFR3 启动子区域,调控其转录表达。FGFR3 敲除或 FGFR 抑制剂(futibatinib)处理,可显著抑制治疗诱导的 DEFA5、DEFA6 表达,阻断类潘氏转化。FGFR3 抑制可降低类潘氏细胞中 pERK 水平,恢复其对双靶向治疗的敏感性;体内 iKAP 模型中,MRTX1133 + 西妥昔单抗联合 futibatinib 可使肿瘤完全逆转为良性腺瘤。
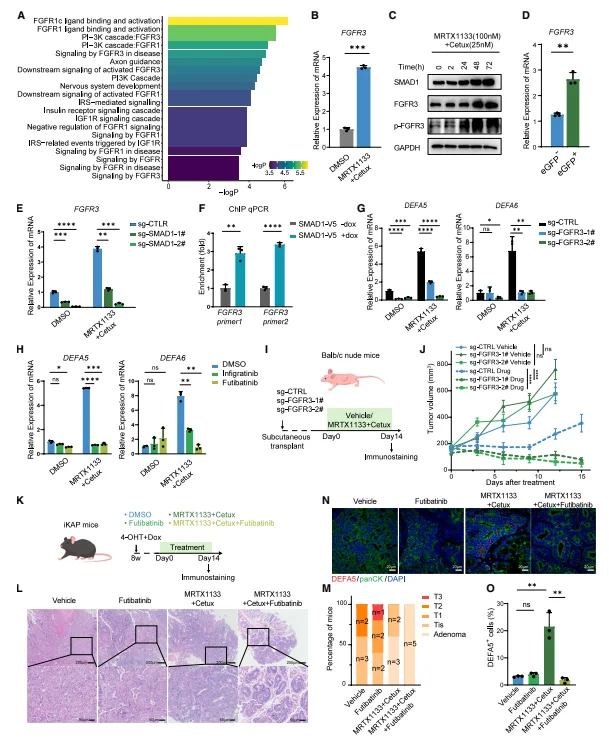
图5. SMAD1-FGFR3信号轴驱动类潘氏细胞状态转换并促进类潘氏细胞中 MAPK 的再激活
-
联合抑制 FGFR3 逆转临床相关模型的耐药 KRAS G12D PDX 模型中,双靶向治疗仅短暂稳定肿瘤,而联合 futibatinib 可诱导肿瘤持续退缩,且显著降低 DEFA5 + 类潘氏细胞比例。临床样本验证:2 例接受 KRAS/EGFR 双靶向治疗的患者,治疗后残留肿瘤中 DEFA5 + 类潘氏细胞显著富集,证实该耐药机制在人类 CRC 中保守存在。
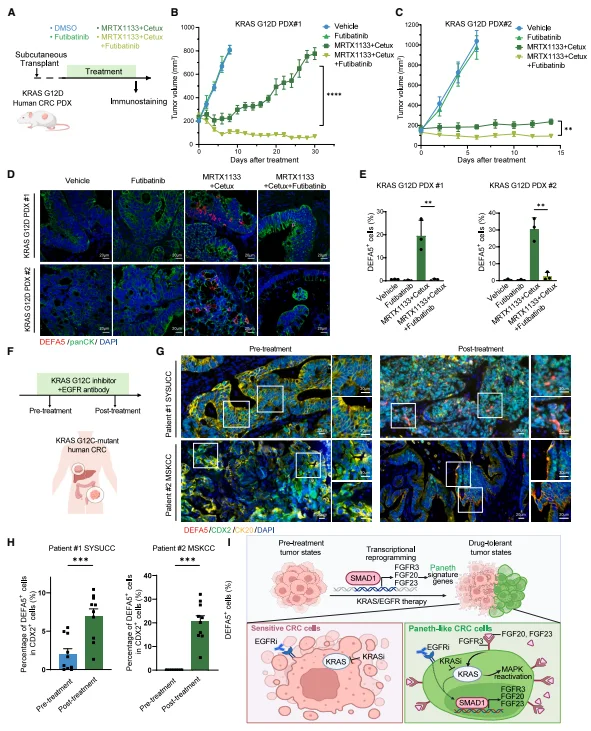
图6.联合KRAS- EGFR 抑制后人类残留结直肠癌肿瘤中类潘氏细胞状态的富集
文章小结
本研究通过多层面实验模型及临床样本,系统证实类潘氏转化是 KRAS 突变 CRC 对 KRAS/EGFR 双靶向治疗产生耐药的核心机制,而 SMAD1-FGFR3 信号轴是驱动该谱系可塑性及 MAPK 通路再激活的关键。临床前数据表明,联合抑制 FGFR3 可有效阻断类潘氏转化,恢复肿瘤对双靶向治疗的敏感性,且该机制在人类 CRC 中保守存在。该研究不仅揭示了 CRC 耐药的全新非遗传机制,更提供了可直接转化的临床治疗方案,有望显著改善 KRAS 突变结直肠癌患者的治疗效果和预后。
源井生物CRISPR-iScreen™文库
CRISPR-iScreen™ 是源井生物自主研发的一项创新技术,旨在实现高效的CRISPR筛选。目前,源井生物拥有超过40种的 CRISPR文库现货 供应,覆盖人类、小鼠、绿猴及猪等物种的全基因组基因敲除/抑制/激活文库;此外,还有针对激酶、细胞周期、膜蛋白、代谢等相关基因构建的CRISPR敲除/抑制/激活亚文库,助您轻松实现靶点筛选!目前源井拥有150+文库病毒现货,400+文库Cell Pool现货,随时支持您的研究需求。
源井生物还提供一站式CRISPR文库(体内/体外)功能筛选服务,搭配全新 iScreenAnlys™ 文库分析平台 Drug-Z/MLE (MAGeCK-MLE)/RRA (MAGeCK-RRA)三大算法自由选,筛选数据一键分析,欢迎咨询加速您的研究进程!
联系我们了解更多>>>参考文献
Zhang Y, Chen J, She Y, Fang Z, Zhang Y, Ruan D, Guo W, Liao J, Zhou W, Lao J, Fang W, Pan X, Kang W, Wang Z, Wu Y, Deng R, Tian L, Wang L, Huang H, Zheng J, Yan Y, Lu H, Wang R, Yaeger R, Zhao Q, Liao W, Wang F, Gao Y. Paneth-like transition drives resistance to dual targeting of KRAS and EGFR in colorectal cancer. Cancer Cell. 2025 Nov 13:S1535-6108(25)00451-9. doi: 10.1016/j.ccell.2025.10.010. Epub ahead of print. PMID: 41237766.







