【前沿盘点】如何利用基因敲除细胞模型把p53基因玩出新花样?

【前沿盘点】如何利用基因敲除细胞模型把p53基因玩出新花样?
TP53基因家族在多细胞动物的发育过程中已经进化了6-8亿年。自该基因家族被发现以来,人们已经花了40年的时间来理解为什么TP53基因是人类癌症中最常见的突变基因。进化上保守的p53蛋白及其细胞通路通过一组对环境扰动起到知情、调节及综合反应来介导对肿瘤的抑制,导致肿瘤细胞死亡或维持细胞内稳态。肿瘤抑制因子p53在细胞应激反应中起着核心作用。p53主要作为转录因子,激活或者抑制多种下游靶基因的转录来行使功能。这些靶基因的作用主要包括诱导细胞周期停滞、DNA修复、细胞衰老、细胞凋亡以及新近发现的诱导细胞发生铁死亡(ferroptosis)等。近年来,随着科学家们研究精力的持续注入,p53基因的神秘面纱被层层揭开。
基因编辑细胞系是科研工作者的科研利器,利用CRISPR/Cas9构建基因编辑细胞系是研究特定基因功能最为常用的技术手段。本文就近期以来,以p53基因编辑细胞系作为研究工具实现对明星抑癌基因p53的突破性研究进行盘点整理,让你轻松走在科研最前沿!
Ferroptosis as a mechanism to mediate p53 function in tumor radiosensitivity(Oncogene,IF=9.8664(2021))
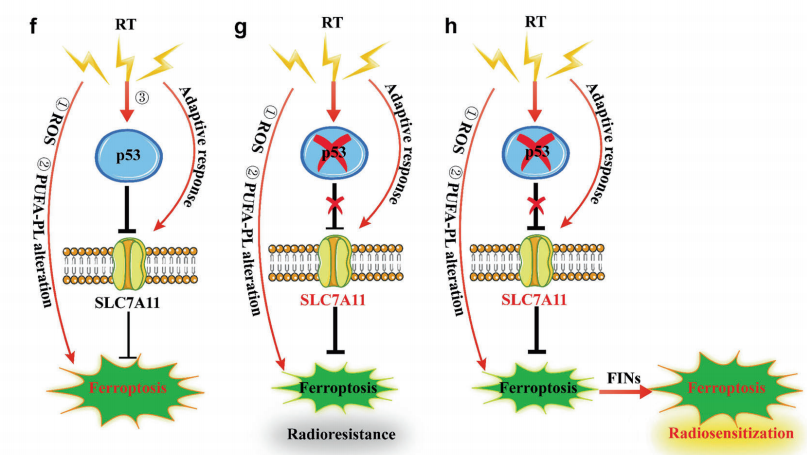
近期,美国德克萨斯大学MD安德森癌症中心的甘波谊教授团队在Oncogene期刊发表题为“Ferroptosis as a mechanism to mediate p53 function in tumor radiosensitivity”,确定了铁死亡是p53在调节肿瘤放射敏感性的关键机制。铁死亡在放射治疗 (RT)诱导的细胞死亡和体内外肿瘤抑 制中发挥重要作用,但是其中的确切遗传背景仍不清楚。鉴于p53是人类癌症中最常见的突变基因,且RT与铁死亡均与p53相关,研究人员就此展开研究,通过构建p53敲除的H1299、A549细胞、过表达p53的A549细胞以及p53敲除肿瘤类器官等细胞系作为研究模型,以确定p53是否调控RT诱导的铁死亡。
最终作者发现,RT介导的p53活化拮抗RT诱导的SLC7A11表达,抑制谷胱甘肽合成,从而促进RT诱导的脂质过氧化和铁死亡。p53缺陷至少部分通过SLC7A11介导的铁死亡抑制促进癌细胞或肿瘤的放射抵抗性。抑制SLC7A11的铁死亡诱导剂(FINs)在p53突变或缺陷的类肿瘤器官和患者来源的异种移植物中发挥显著的放射增敏作用。另外,作者发现了RT诱导的铁死亡与p53激活和癌症患者RT更好的临床结果相关。总之,该研究揭示了铁死亡在p53介导的放射增敏中尚未引起重视的作用,并建议使用FINs联合RT治疗p53突变的癌症。
想构建p53基因敲除细胞?点此进入源井生物独家KO细胞库即可现货一周达 >> 近8000种KO细胞产品包含p53敲除A549、HEK293细胞系等低至4980,覆盖上千种基因满足各类科研需求!
Acquisition of aneuploidy drives mutant p53-associated gain-of-function phenotypes(Nature Communications,IF=14.9196(2021))
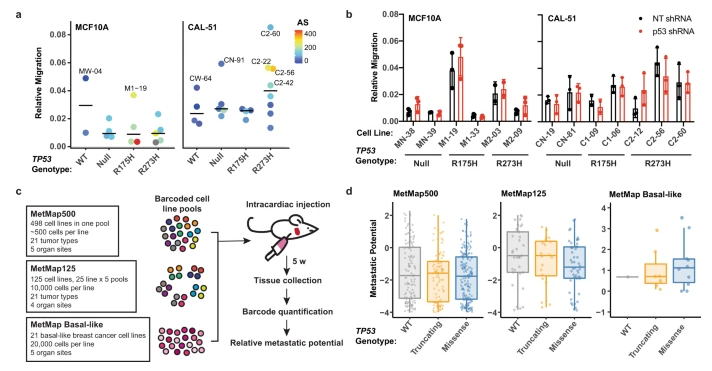
在超过一半的人类癌症中均检测到存在p53基因的突变。p53基因的突变除了失去肿瘤抑制功能外,还有可能促进肿瘤的发生与发展,从而导致新的致癌表型,这一现象被称为功能获得性突变(gain of function,GOF)。在研究这种GOF发生机制和表型的过程中,来自范德堡大学医学中心(Vanderbilt University Medical Center)的研究人员发现,在p53突变的细胞中,驱动GOF的是一种称为非整倍体的现象,而与编码p53的TP53基因型无关。这不仅推翻了已有的研究结论,同时对于开发针对p53突变的创新疗法具有启示意义。
在这篇文章中,为了研究突变 p53 GOF机制和表型,研究人员对非转化和肿瘤衍生的两种细胞系模型(即MCF10A细胞系和CAL-51细胞系)进行了CRISPR/Cas9基因组编辑,以表达常见的几种p53突变蛋白(包括WT、Null、R175H和R273H),最终总共生成36种不同细胞系模型。结果发现,MCF10A和CAL-51这两种基因编辑细胞系均表现为非整倍体出现频率显著增加。进一步研究发现,体外GOF表型仅存在于非整倍性表达增加的p53突变表达细胞系中,且这些表型不依赖于突变p53蛋白表达。总之这一系列试验均表明,与基因表达的变化、肿瘤形成和转移密切相关的是非整倍体,而非p53突变。这项研究揭示了p53突变导致癌症发生的新机制,为开发针对突变p53的疗法带来了崭新的思路。
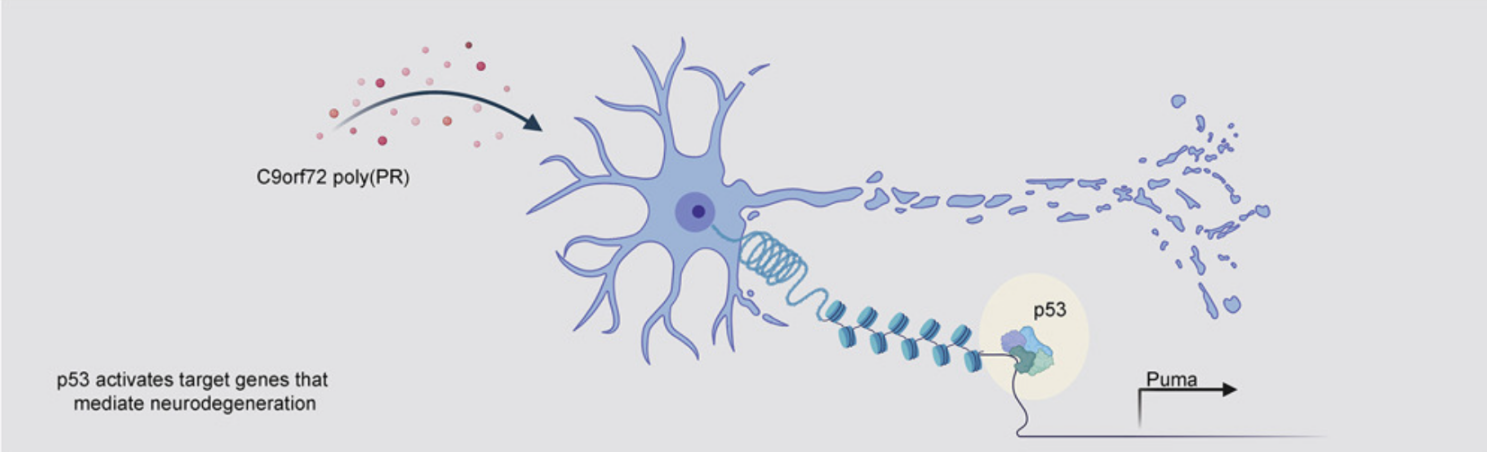
p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR) (Cell, IF=41.5836(2021))
肌萎缩侧索硬化症 (ALS) 和额颞叶痴呆 (FTD) 最常见的遗传原因是 C9orf72 基因中的 GGGGCC 重复扩增。近期,来自美国斯坦福大学医学院的Aaron D. Gitler和Maya Maor-Nof在Cell杂志上合作发表了一篇题为p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR) 的文章。在这项研究中,作者开发一种作用于原代神经元的改良型ATAC-seq,揭示了p53在poly(PR)诱导的神经退行性变中发挥的意想不到的作用,并提供了一个将ATAC-seq应用于神经元来定义神经退行性变机制的范例。为了研究C9orf72六核苷酸重复序列扩增产生的两种ALS相关蛋白TDP-43和poly(PR)聚集体是通过相似还是不同的机制导致变性,作者构建了一个体外系统,用于监测由poly(PR)或TDP-43累积引起的神经元死亡和轴突变性。具体的,将小鼠原代脑皮层神经元在体外培养,转以分别表达 (PR)50、TDP-43或GFP的慢病毒,在72h左右观察到轴突变性,神经元死亡等现象。进一步对神经元提取的DNA及RNA进行后续的ATAC-seq和RNA-seq发现,p53可能是C9orf72(PR)50引起神经退行性变的一个中心调节因子。
为了验证这一发现,作者对p53-KO和p53-WT小鼠的原代神经元转以表达TDP-43或(PR)50的慢病毒,得出p53-KO神经元可持久性的免受(PR)50而非TDP-43引起的神经变性的影响。进一步,作者使用腺相关病毒载体构建的转基因小鼠模型,在小鼠大脑中表达C9orf72(PR)50,并观察这些小鼠的存活情况以确定p53- KO在体内也能免受神经变性的保护。为了研究p53是否参与人类模型中C9orf72-GGGGCC重复扩增的发病机制,研究人员还使用了来自C9orf72-ALS患者的iPSC,且在iPSC分化的运动神经元中发现DNA损伤增加。在使用shRNA部分减少p53后可观察到DNA损伤程度减轻,提示p53在C9orf72阳性神经元中被激活,诱导DNA损伤和细胞凋亡机制。
总的来说,这项研究通过ATAC-seq在原代神经元中的应用,并结合小鼠模型和临床患者来源的iPSC实验数据,揭示了p53在C9orf72(PR)50诱导的神经元退行性变中的作用。
P53基因自从在1979年被首次报道以来,有关研究论文在Medline上可查到数万篇,数十年的研究似乎已经让我们对其具有充分的了解,然而随着生物科技领域高速的更新迭代,如CRISPR/Cas9基因编辑技术的出现与基因编辑细胞模型的精准构建让我们能更高效、更可控地对P53基因进行更多的探索,甚至会推翻过往的认知。
源井生物基于传统的CRISPR/Cas9技术进一步研发了CRISPR-U™独家专利技术,具有更高的基因编辑效率,目前源井生物已凭借该技术成功为全球20多个国家与地区提供了数千次基因编辑细胞服务,并成功构建了拥有近8000+KO细胞的基因敲除细胞库,现低至4980即可一周内获得成功敲除的KO细胞,轻松实现各类研究。






