EMBO J | FOXP1 通过磷酸化与O -糖基化的拮抗作用调控ATR激活及复制应激应答

ATR信号通路是细胞应对复制压力的核心防线,其激活异常会导致基因组不稳定,进而引发肿瘤等疾病。FOXP1作为叉头框家族转录因子,此前被认为主要参与转录调控,深圳大学医学部许兴智教授团队与深圳大学总医院巩鹏教授团队联合发表在《THE EMBO JOURNAL》期刊上的文章显示,FOXP1以支架蛋白身份,通过直接结合RPA-ssDNA复合物与ATR-ATRIP复合物,介导ATR的招募与激活。更关键的是,FOXP1的功能受O-糖基化(O-GlcNAcylation)与磷酸化的双重调控:OGT介导的O-糖基化抑制其与ATR的相互作用,而复制压力下CHK1介导的S396位点磷酸化会拮抗该糖基化修饰,形成正向调控环路。肿瘤组织中FOXP1的致病性突变会破坏这一机制,导致ATR激活缺陷和复制叉稳定性下降。 源井生物为该研究提供了FOXP1基因S396A和S396D点突变HEK293细胞系,助力研究揭示FOXP1在DNA损伤应答中的非转录功能,为相关肿瘤的靶向治疗提供了新的分子靶点。

研究背景
DNA复制过程常面临内源性(如dNTP合成异常)和外源性(如紫外线、化疗药物)压力,导致复制叉停滞,若不能及时修复会引发DNA断裂和基因组不稳定。ATR-CHK1通路是应对复制压力的关键信号网络,ATR激酶的激活需通过与RPA-ssDNA复合物结合,并在TopBP1、ETAA1等激活因子作用下实现,其过程受磷酸化、泛素化等翻译后修饰(PTMs)的精密调控。O-糖基化作为另一重要PTM,与磷酸化共享丝氨酸/苏氨酸修饰位点,二者常存在竞争或协同关系,但在ATR信号通路中的作用尚未明确。FOXP1作为多功能转录因子,其单倍体不足与发育迟缓、肿瘤发生相关,且症状与ATR信号缺陷相似,但FOXP1是否参与复制压力应答及其调控机制仍未知。
研究目的
明确FOXP1在ATR激活及复制压力应答中的功能,解析其O-糖基化与磷酸化修饰的调控机制,阐明FOXP1致病性突变影响复制压力应答的分子机理,为肿瘤治疗提供潜在靶点。
研究方法
细胞与分子实验: 使用HEK293T、H1975等细胞系,通过免疫沉淀(IP)、GSTpull-down、蛋白质印迹(IB)验证蛋白相互作用;利用siRNA敲低、CRISPR-Cas9敲入技术调控FOXP1表达及突变;通过体外磷酸化实验、O-糖基化检测验证翻译后修饰。
功能验证实验: 采用DNA纤维实验检测复制叉稳定性;细胞周期分析评估S期阻滞;细胞活力实验检测对复制压力药物(HU、CPT)的敏感性;免疫荧光、邻近连接实验(PLA)、iPOND技术检测FOXP1在复制叉的定位。
突变体分析: 构建FOXP1致病性突变体(R465G、R514C等)及修饰位点突变体(S396A、S396D等),验证其对蛋白相互作用、ATR激活及复制叉稳定性的影响。
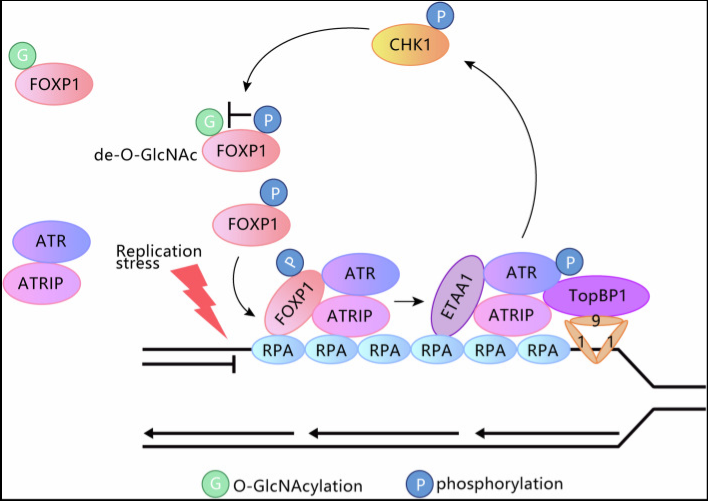
图1、FOXP1在复制应激反应中的功能工作模型
研究路线
筛选与验证: 通过ATR抗体IP-MS筛选相互作用蛋白,鉴定FOXP1;验证FOXP1与ATR-ATRIP、RPA-ssDNA的相互作用,及复制压力对该相互作用的增强效应。
功能确认: 敲低FOXP1检测ATR激活(CHK1S345磷酸化)、复制叉稳定性及细胞对复制压力的敏感性,明确FOXP1对ATR通路的正向调控作用。
修饰机制解析: 验证OGT介导FOXP1的O-糖基化及其对ATR相互作用的抑制;鉴定CHK1介导FOXP1的S396位点磷酸化,及其对O-糖基化的拮抗作用。
突变体功能分析: 检测肿瘤相关FOXP1突变体对DNA结合、蛋白相互作用、ATR激活及复制叉稳定性的影响,揭示突变体的致病机制。
机制整合: 构建FOXP1致病性突变体(R465G、R514C等)及修饰位点突变体(S396A、S396D等),验证其对蛋白相互作用、ATR激活及复制叉稳定性的影响。
主要结果
FOXP1促进ATR激活: FOXP1与ATR-ATRIP复合物直接相互作用,且该相互作用在HU诱导的复制压力下增强;敲低FOXP1显著抑制ATR介导的CHK1S345磷酸化,降低复制叉稳定性,增强细胞对HU的敏感性。
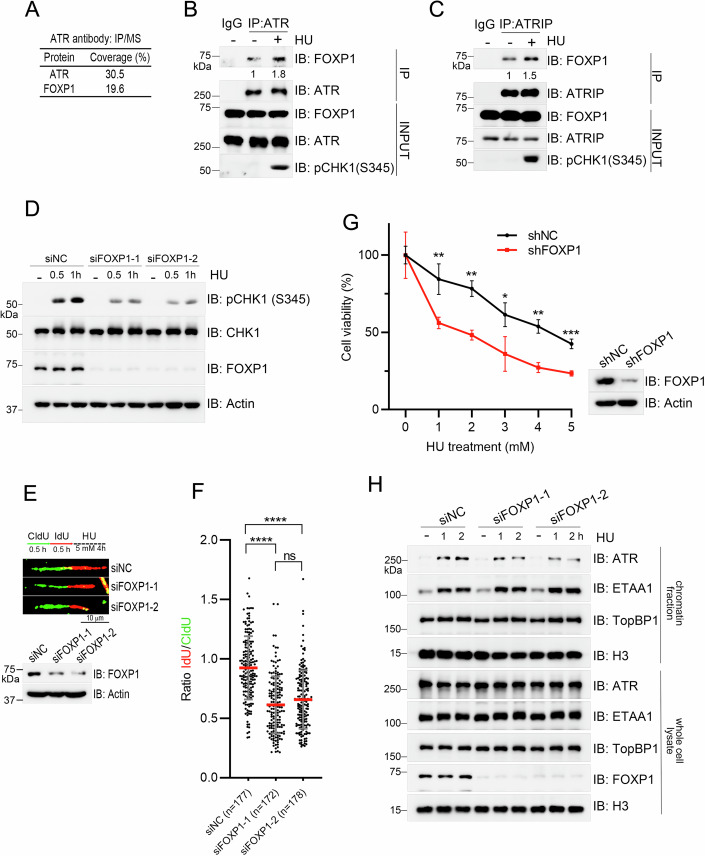
图2、FOXP1在复制应激条件下促进ATR激活
FOXP1定位于停滞复制叉: FOXP1通过叉头结构域(465-555AA)直接结合ssDNA,且对RPA包被的ssDNA亲和力更高;FOXP1与RPA70/RPA32直接相互作用,复制压力下通过RPA依赖的方式募集到停滞复制叉。
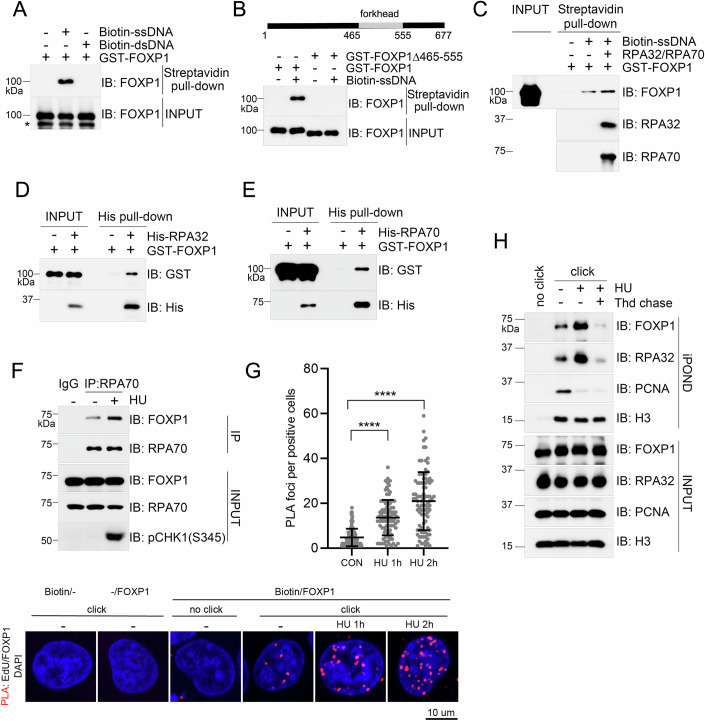
图3、FOXP1结合于停滞的复制叉上
O-糖基化抑制FOXP1-ATR相互作用: OGT直接催化FOXP1的O-糖基化(依赖556-610AA结构域);复制压力下FOXP1的O-糖基化水平下降,O-糖基化修饰会抑制FOXP1与ATR的结合。
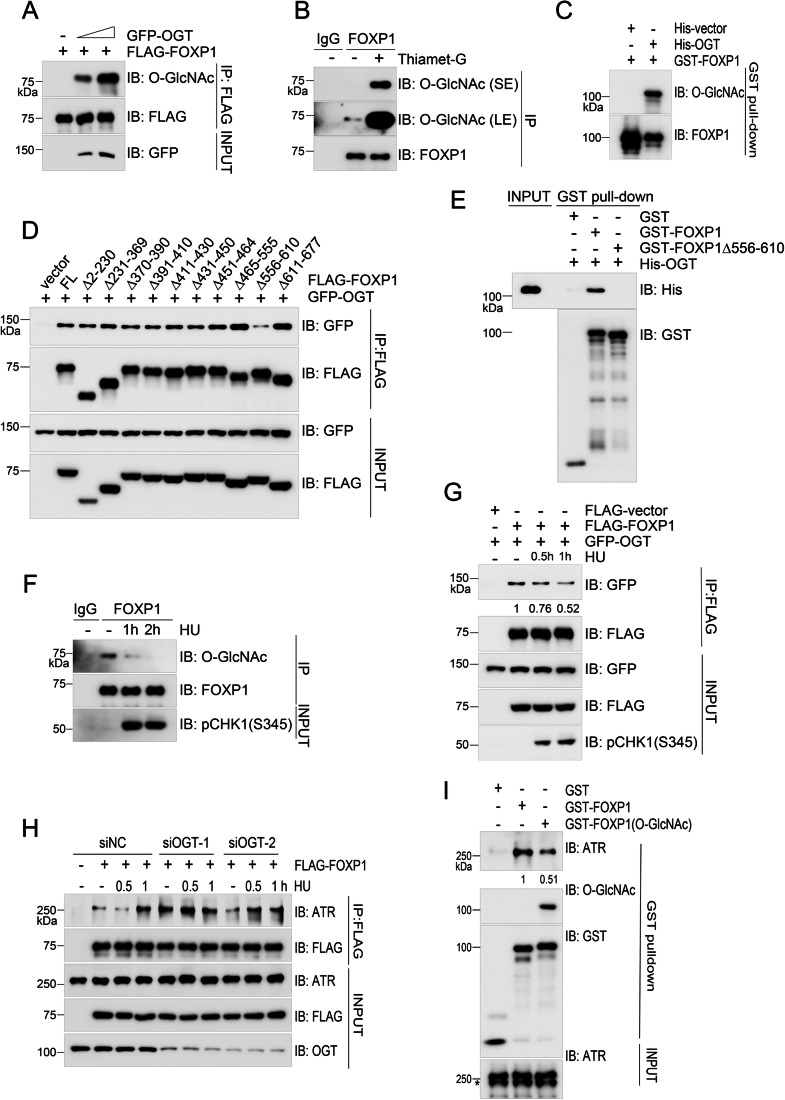
图4、OGT介导的FOXP1O-GlcNAcylation修饰抑制其与ATR的相互作用
CHK1介导磷酸化拮抗O-糖基化: CHK1直接磷酸化FOXP1的S396位点;磷酸化突变体S396A的O-糖基化水平升高,与ATR的相互作用减弱,ATR激活缺陷;磷酸模拟突变体S396D则相反,且能挽救ATR激活缺陷。
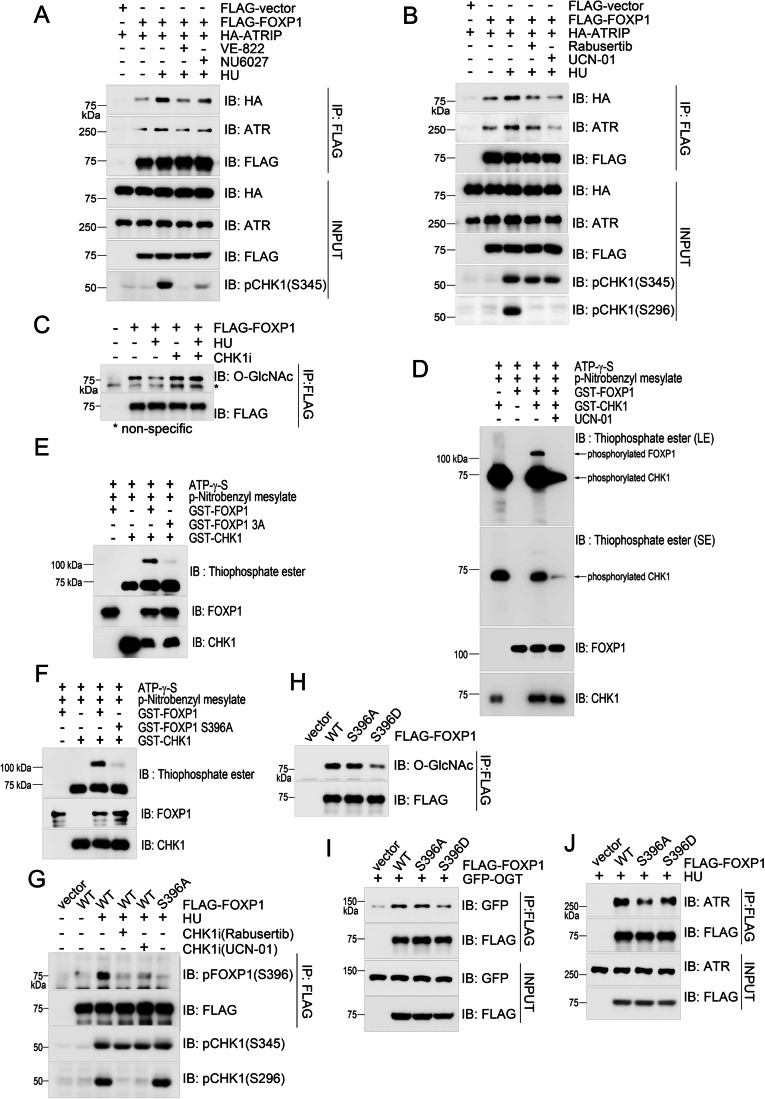
图5、CHK1介导的FOXP1在S396位点的磷酸化作用与其O-GlcNAcylation过程相互拮抗
FOXP1突变体功能异常: 肿瘤相关突变体(R465G、R514C等)降低FOXP1与ssDNA、RPA的结合能力,增强O-糖基化水平,减弱与ATR的相互作用,最终导致ATR激活不足和复制叉稳定性下降。
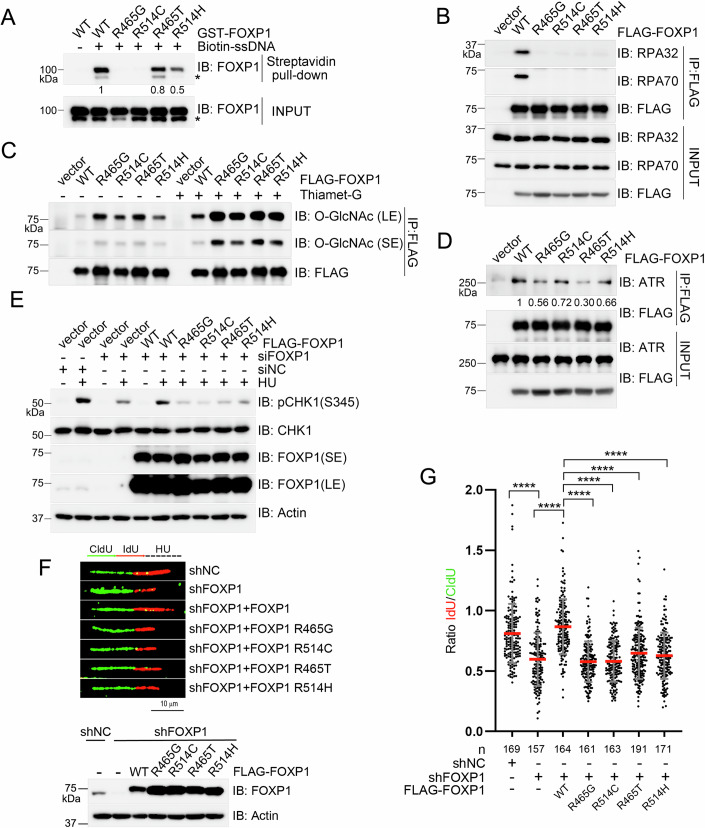
图6、致病性FOXP1突变在复制应激条件下会损害其功能
研究意义与创新点
理论创新: 首次发现FOXP1的非转录功能,即作为支架蛋白介导ATR招募与激活,拓展了FOXP1的生物学功能边界;揭示O-糖基化与磷酸化的相互拮抗在ATR通路调控中的关键作用,完善了复制压力应答的PTM调控网络。
机制突破: 阐明“CHK1-FOXP1(磷酸化)-OGT-FOXP1(O-糖基化)-ATR”的正向调控环路,为理解ATR激活的精密调控机制提供了新视角。
临床价值: 明确肿瘤相关FOXP1突变体通过破坏复制压力应答促进癌变,为肿瘤分型提供分子标志物;FOXP1作为ATR激活的关键调控因子,有望成为肿瘤化疗(复制压力诱导剂)的联合治疗靶点。
文章小结
本研究首次揭示了转录因子FOXP1在复制压力应答中的非转录功能,即通过支架蛋白活性介导ATR的招募与激活。FOXP1的功能受双重翻译后修饰调控:OGT介导的O-糖基化抑制其与ATR的相互作用,而复制压力下CHK1介导的S396位点磷酸化会拮抗该糖基化修饰,从而增强FOXP1对ATR的招募,保障ATR通路激活和复制叉稳定。肿瘤组织中FOXP1的致病性突变会破坏其DNA结合、蛋白相互作用及修饰平衡,导致ATR激活缺陷,进而促进基因组不稳定和肿瘤发生。该研究不仅完善了ATR信号通路的调控网络,还为FOXP1相关肿瘤的诊断和靶向治疗提供了重要的理论依据和潜在靶点。
源井生物提供的支持
该研究选用源井生物定制的 FOXP1 基因 S396A 、 S396D 点突变 HEK293 细胞系,为解析 FOXP1 致病性突变调控复制应激应答的分子机制提供了关键实验模型支撑。
依托多年深耕基因编辑领域的技术积淀,源井生物已累计完成超 13000 例基因编辑成功案例,构建起成熟稳定的技术服务体系。针对 点突变细胞构建需求 源井提供一站式解决方案, 涵盖 RNP 法、单碱基编辑器、先导编辑器、质粒抗性法 4 种技术路径, 可灵活适配不同突变场景,低至 1.58 w轻松解放双手。凭借自主创新研发的 EZ-HRex™技术,HDR基因型占比最高可达 84%,为科研人员提供高效、精准的基因编辑工具支持。
参考文献
ZhuX,GaoC,PengB,XueJ,XiaD,YangL,ZhangJ,GaoX,HuY,LinS,GongP,XuX.FOXP1phosphorylationantagonizesitsO-GlcNAcylationinregulatingATRactivationinresponsetoreplicationstress.EMBOJ.2025Jan;44(2):457-483.doi:10.1038/s44318-024-00323-x.Epub2024Dec2.PMID:39623140;PMCID:PMC11729909.







