CRISPR 筛选鉴定 JUN 为 SHP2/ERK 与 RAS(ON) 抑制剂共性耐药靶点

引言
胰腺癌是全球致死率最高的恶性肿瘤之一,KRAS 突变在胰腺导管腺癌(PDAC)中发生率超 85%,是核心致癌驱动因素。针对 MAPK 通路的联合靶向治疗(如 SHP2+ERK 抑制剂)已进入临床研究阶段,但耐药问题仍是制约疗效的关键。荷兰癌症研究所的Rene Bernards与Sara Mainardi团队通过 全基因组CRISPR敲除筛选 及后续功能验证,首次明确 PI3K/AKT/mTOR 通路激活与 JUN 高表达是 KRAS 突变 PDAC 对 MAPK 抑制耐药的相互关联机制,且 JUN 是最下游的核心耐药介导因子;同时证实靶向 MAP2K4 抑制 JUN 可有效逆转耐药,为优化 KRAS 突变胰腺癌的联合治疗策略提供了重要理论依据和潜在靶点。

研究背景
胰腺癌 5 年生存率仅 10%,晚期诊断和治疗手段匮乏是主要原因,传统化疗疗效有限,靶向治疗仅对极少数同源重组缺陷患者有效。KRAS 作为胰腺癌最主要的致癌驱动基因,曾因结构特性被认为 “不可成药”,近年来虽开发出 KRASG12C 特异性抑制剂并获批用于非小细胞肺癌,但胰腺癌中最常见的 KRASG12D 突变抑制剂仍处于早期临床阶段。目前,SHP2+ERK 或 RAS (ON) 多选择性抑制剂联合治疗的耐药机制尚未明确,亟需系统解析耐药相关分子及通路,为临床制定应对策略提供支撑。
研究目的
系统鉴定 KRAS 突变 PDAC 对 SHP2+ERK 抑制剂及 SHP2+RAS (ON) 多选择性抑制剂联合治疗的耐药介导因子,阐明其分子机制,验证靶向干预耐药通路的可行性,为 KRAS 突变胰腺癌的精准治疗提供新靶点和策略。
研究方法
-
细胞模型构建:
- 选用 YAPC-1、ASPC-1、Panc 10.05、Panc-1、MiaPaCa-2 等 KRAS 突变 PDAC 细胞系,通过持续暴露于 SHP2+ERK 抑制剂(RMC-4550+LY3214996)或 SHP2+RAS (ON) 抑制剂(RMC-4550+RMC-6236)构建 MiaPaCa-2 自发耐药细胞系(Resistant、Resistant_1)。
- 通过慢病毒转导 sgRNA 构建 PTEN 敲除单克隆细胞系,通过慢病毒转导过表达载体构建 JUN 过表达细胞系;所有细胞经 STR 鉴定和支原体检测,HEK293T 细胞用于慢病毒包装。
- 全基因组 CRISPR 敲除筛选: 在 Panc 10.05 细胞中感染 Brunello 全基因组 sgRNA 文库,经嘌呤霉素筛选后分为低剂量(2μM RMC-4550+2μM LY3214996)、高剂量(4μM RMC-4550+4μM LY3214996)处理组和未处理组,培养后回收 sgRNA 并测序,通过 DESeq2 和 MAGeCK RRA 分析筛选耐药相关基因。
- 聚焦 CRISPR 筛选: 针对初筛得到的 167 个基因设计定制化 sgRNA 文库,在 4 种 KRAS 突变 PDAC 细胞系中进行高覆盖率(800×)筛选,结合 STRING 软件进行互作组分析,锁定核心候选基因。
- 分子生物学实验: 利用WB、IHC验证通路激活状态和情况。
- 动物实验: 将 Panc 10.05 PTEN 敲除细胞(5×10⁶个)接种于 NSG 小鼠右侧胁腹,构建异种移植瘤模型,待肿瘤体积达 200-250 mm³ 时,分为溶剂组、HRX-0233 单药组、SHP2+ERK 抑制剂联用组、三联用药组(SHP2+ERK 抑制剂 + HRX-0233),通过灌胃给药,监测肿瘤体积变化,验证体内靶向干预效果。
研究路线
- 筛选耐药介导因子: 通过两步法 CRISPR 敲除筛选(全基因组 + 聚焦),在 KRAS 突变 PDAC 中鉴定出 PTEN、DET1、COP1 等耐药相关基因,互作组分析证实 PTEN 为核心枢纽基因。
- 验证 PTEN 敲除的耐药作用: 构建 PTEN 敲除细胞系,证实其通过激活 PI3K-AKT-mTOR 通路产生对 SHP2+ERK 抑制剂的耐药,且自发耐药细胞模型也存在该通路激活,验证该机制的普遍性。
- 解析 mTOR 通路与 JUN 的关联: 发现 PTEN 敲除 / 自发耐药细胞中 JUN 高表达,证实 mTOR 通路激活可驱动 JUN 表达,且 JUN 是 mTOR 下游的关键分子。
- 验证 JUN 为核心耐药介导因子: 通过 JUN 过表达实验证实其可独立介导耐药,通过靶向抑制 JNK/MAP2K4 下调 JUN,可恢复细胞对 SHP2+ERK 抑制剂的敏感性。
- 体内验证逆转耐药的策略: 在 PTEN 敲除异种移植瘤模型中,证实 MAP2K4 抑制剂 HRX-0233 与 SHP2+ERK 抑制剂联用可有效抑制肿瘤生长,逆转体内耐药。
- 拓展 JUN 的耐药介导作用: 证实 JUN 同样介导 KRAS 突变 PDAC 对 SHP2+RAS (ON) 多选择性抑制剂联合治疗的耐药,明确其为广谱的耐药核心节点。
主要结果
1.CRISPR 基因筛选揭示了KRAS突变 PDAC 中对SHP2联合ERK抑制耐药的中介因子
研究团队首先在 Panc 10.05 细胞中开展 全基因组 CRISPR 筛选, 以不同剂量的 SHP2+ERK 抑制剂组合作为筛选压力,最终鉴定出多个耐药相关基因。其中,低、高剂量处理组共有的核心基因包括 PTEN、PPP6C、PPP2R4 等 6 个,而高剂量组因筛选条件更严格,鉴定出的基因数量相对更少。为进一步细化研究结果,团队在 4 种 KRAS 突变 PDAC 细胞系中进行 聚焦 CRISPR 筛选, 发现耐药基因存在明显的细胞系特异性:MiaPaCa-2 和 ASPC-1 细胞中共同的耐药基因为 DET1 和 COP1,二者可形成 E3 泛素连接酶复合物,进而调控 JUN 蛋白稳定性;Panc 10.05 和 Panc-1 细胞则共享 PTEN、CYB5R4 等耐药基因;PPP2R4 虽在 3 种细胞系中均有富集,但经 sgRNA 单独验证后,未证实其具备耐药调控作用。后续通过 STRING 互作组分析进一步发现,PTEN 是所有筛选所得耐药基因中的核心枢纽分子,这也使其成为后续机制深入探究的核心候选基因。
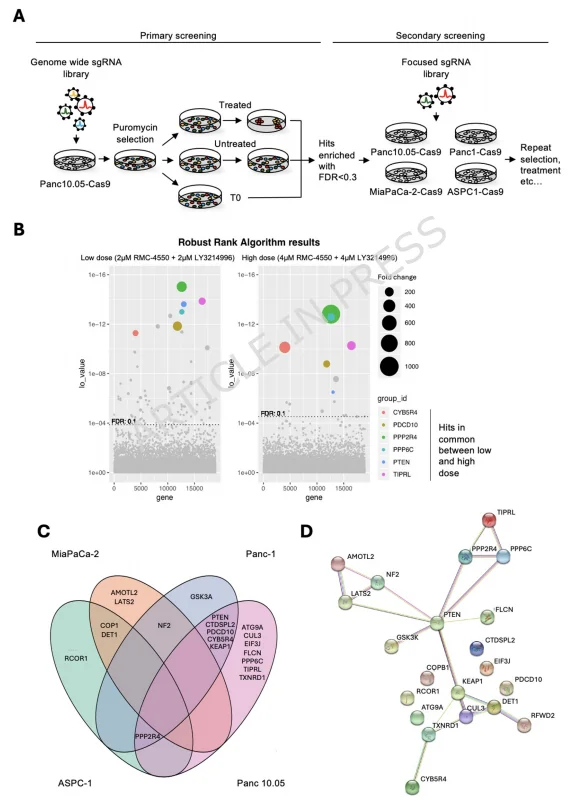
图1.CRISPR 基因筛选揭示了KRAS突变 PDAC 中对SHP2联合ERK抑制耐药的中介因子
2.PTEN 敲除或 PI3K-AKT-mTOR 通路自发激活介导对 SHP2+ERK 抑制剂的耐药
接着,研究团队发现在PTEN 敲除细胞系(Panc 10.05、Panc-1、ASPC-1)中,PI3K-AKT-mTOR 通路激活(p-AKT、p-S6RP 升高),对 SHP2+ERK 抑制剂的耐药性显著增加,且药物诱导的细胞凋亡被抑制。为进一步验证耐药机制的普遍性,研究构建了 MiaPaCa-2 自发耐药细胞系。结果显示,其未出现 MAPK 通路复激活(p-RSK 水平低),但 PTEN 表达降低、p-S6RP 升高,与 PTEN 敲除细胞具有相似的信号特征,且耐药细胞存在 “药物成瘾” 现象,撤药后增殖受抑。自发耐药细胞的单克隆株均保留耐药性,且均存在 PI3K-AKT-mTOR 通路激活;MiaPaCa-2 异种移植瘤经 SHP2+ERK 抑制剂处理后,p-S6RP 染色增强,证实体内治疗过程中 mTOR 通路激活参与获得性耐药。mTOR 抑制剂 AZD8055 可抑制耐药细胞增殖,与 SHP2+ERK 抑制剂联用可有效抑制亲本和耐药细胞的生长,但对部分细胞系(MiaPaCa-2、Panc-1)的耐药预防效果不完全,提示存在 mTOR 非依赖的耐药机制。
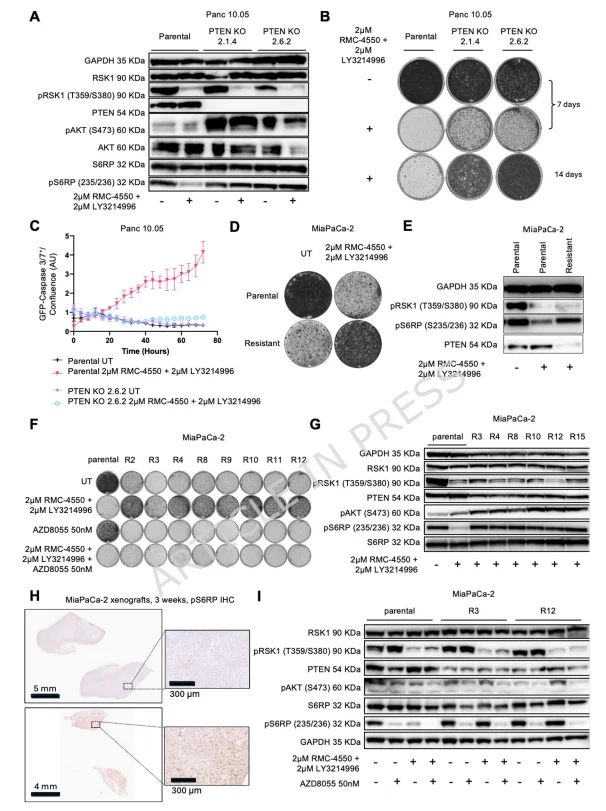
图2.PTEN 敲除或 PI3K-AKT-mTOR 通路自发激活介导对 SHP2+ERK 抑制剂的耐药
3.JUN 是 KRAS 突变 PDAC 中 MAPK 抑制耐药的最下游介导因子
团队进一步研究发现,在MiaPaCa-2 自发耐药细胞克隆及 PTEN 敲除细胞中,总 JUN 和磷酸化 JUN(p-JUN)水平显著升高,且不受 SHP2+ERK 抑制剂抑制,而亲本细胞中 p-JUN 水平随药物处理降低。进一步的RNA-seq 分析显示,PTEN 敲除细胞中 AP-1 转录因子网络失调,JUN 显著上调,而抑制性 AP-1 家族成员 BATF 下调,提示 AP-1 活性向促增殖方向转变。
mTOR 抑制剂 AZD8055 可同时下调 mTOR 靶标 S6RP 及 JUN(总 JUN 和 p-JUN)的表达,且 SHP2+ERK 抑制剂与 AZD8055 联用可恢复 PTEN 敲除细胞对药物的敏感性。靶向抑制 JUN 上游分子(JNK 抑制剂 SP600125、MAP2K4 抑制剂 HRX-0233)可降低 p-JUN 水平,恢复 PTEN 敲除细胞对 SHP2+ERK 抑制剂的敏感性;反之,JUN 过表达可直接赋予细胞耐药性,且不依赖 MAPK 通路复激活。
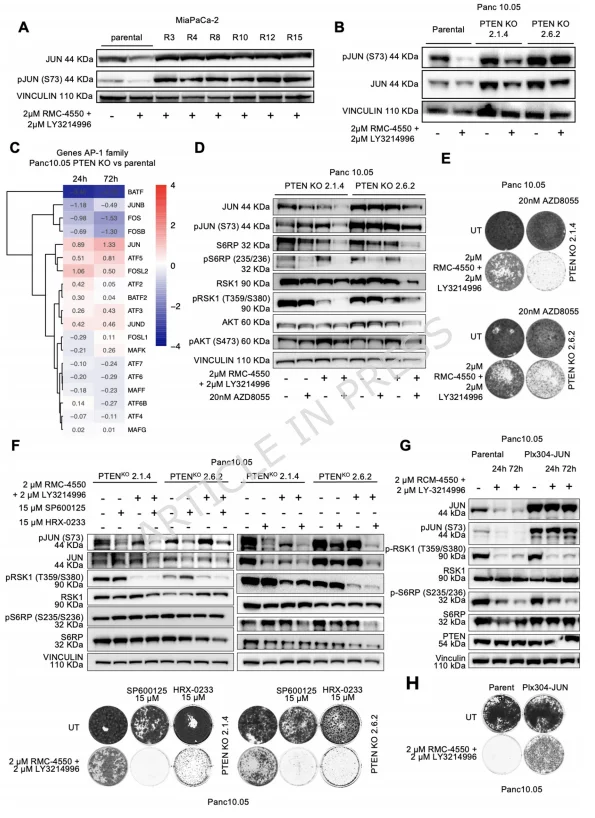
图3.JUN 是 KRAS 突变 PDAC 中 MAPK 抑制耐药的最下游介导因子
4.JUN 为体内耐药靶点,且介导对 SHP2+RAS (ON) 抑制剂的耐药
体内实验显示,Panc 10.05 PTEN KO 异种移植瘤对 SHP2+ERK 抑制剂耐药,而联合 MAP2K4 抑制剂 HRX-0233 可有效抑制肿瘤生长,证实靶向 JUN 可逆转体内耐药。SHP2 抑制剂 RMC-4550 与 RAS (ON) 多选择性抑制剂 RMC-6236 联用在 PDAC 细胞系中具有显著协同抗瘤效果;研究团队通过构建 MiaPaCa-2 自发耐药细胞系(Resistant_1),发现其耐药表型较 SHP2+ERK 抑制剂耐药细胞更温和,且撤药后仍可增殖。Resistant_1 细胞中 PTEN 下调,但 mTOR 通路未激活,而 p-JUN 水平显著升高,提示 JUN 以 mTOR 非依赖方式介导对 SHP2+RAS (ON) 抑制剂的耐药。 JUN 过表达可直接赋予 Panc 10.05、Panc-1 细胞对 SHP2+RAS (ON) 抑制剂的耐药性,且不依赖 MAPK 通路复激活,证实 JUN 是广谱的耐药核心节点。
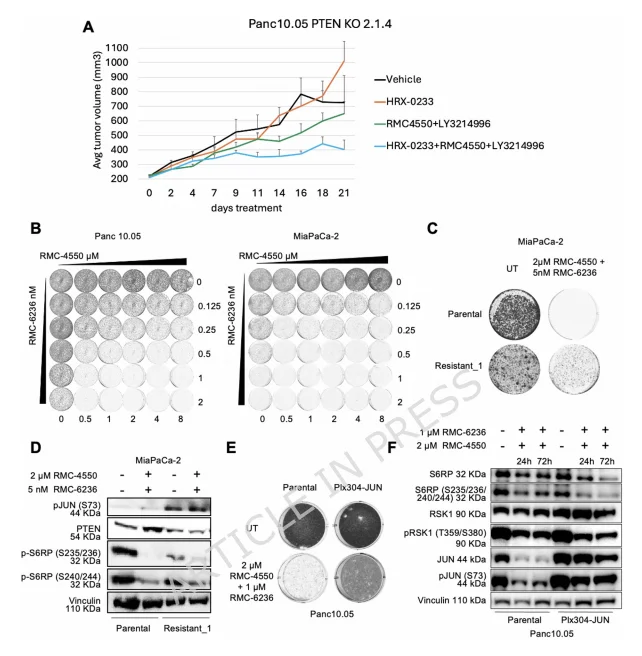
图4.JUN 为体内耐药靶点,且介导对 SHP2+RAS (ON) 抑制剂的耐药
文章小结
本研究通过全基因组 CRISPR 筛选结合体内外功能验证, 系统解析了 KRAS 突变 PDAC 对 MAPK 抑制的耐药机制,证实 PTEN 缺失介导的 PI3K-AKT-mTOR 通路激活与 JUN 高表达是核心耐药机制,且 JUN 是最下游的关键介导因子。 同时,研究明确靶向 MAP2K4 抑制 JUN 可有效逆转耐药,为优化 KRAS 突变胰腺癌的联合治疗策略提供了重要理论依据和潜在靶点。该发现不仅丰富了胰腺癌靶向治疗耐药的分子机制研究,也为临床开发更有效的联合治疗方案、改善患者预后奠定了基础。
源井生物CRISPR-iScreen™文库
CRISPR-iScreen™是源井生物自主研发的一项创新技术,旨在实现高效的CRISPR筛选。目前,源井生物拥有超过40种的CRISPR文库现货供应,覆盖人类、小鼠、绿猴及猪等物种的全基因组基因敲除/抑制/激活文库;此外,还有针对激酶、细胞周期、膜蛋白、代谢等相关基因构建的CRISPR敲除/抑制/激活亚文库,助您轻松实现靶点筛选!目前源井拥有150+文库病毒现货,400+文库Cell Pool现货,还提供一站式CRISPR文库(体内/体外)功能筛选服务,随时支持您的研究需求。
联系我们了解更多>>>参考文献
Mulero-Sánchez A, Bosma A, Visuvasam B, Pouliopoulou N, van de Ven M, Proost N, Boeije M, Lieftink C, Beijersbergen R, Bernards R, Mainardi S. CRISPR knockout screens reveal JUN as the master mediator of resistance to MAPK inhibition in KRAS-mutant pancreatic cancer. J Exp Clin Cancer Res. 2026 Jan 22. doi: 10.1186/s13046-025-03616-z. Epub ahead of print. PMID: 41572361.







