锁定免疫增效开关 KLHL6!CRISPR 筛选驱动 T 细胞疗法突破实体瘤障碍

引言
肿瘤浸润 T 细胞的耗竭与线粒体功能异常是癌症免疫治疗的核心障碍,而蛋白质稳态对这一过程的调控尚未明确。近日,李贵登团队在Nature发表最新研究,通过计算分析结合靶向 体内 CRISPR 筛选 ,鉴定出 E3 泛素连接酶 KLHL6 是两者的双重负调控因子。其机制为 KLHL6 可促进 TOX 泛素化降解以抑制 T 细胞终末耗竭,同时通过调控 PGAM5–Drp1 轴维持线粒体稳态,但 T 细胞受体刺激会下调其表达。强制表达 KLHL6 能显著提升 T 细胞体内抗肿瘤及抗病毒的疗效与存续能力,为癌症免疫治疗提供了新的临床可行靶点。

研究背景
在肿瘤微环境或慢性病毒感染(如LCMV)中,CD8+ T细胞因长期暴露于抗原,其转录谱和代谢模式发生显著改变,最终导致效应功能丧失、抑制性受体(如PD-1)持续高表达。过往研究多聚焦于耗竭状态的表征或寻找逆转耗竭的因子,但对于持续性TCR信号究竟通过何种分子机制驱动了从效应T细胞向耗竭T细胞的转化,即耗竭启动的核心调控节点,缺乏深入的理解。解析这一机制对于开发能够维持T细胞持久功能的治疗策略至关重要。
研究目的
该研究旨在鉴定并解析在持续性TCR信号刺激下,调控CD8+ T细胞从激活转向耗竭的关键分子开关。作者希望通过整合系统生物学与功能基因组学手段,找到能够同时影响T细胞耗竭进程和线粒体代谢状态的核心调控因子,并探索其在肿瘤免疫治疗中的潜在应用价值。
研究方法
生物信息学分析:
利用已有的转录组数据,系统表征和分析T细胞在急性感染与慢性感染过程中基因表达的动态变化,筛选与耗竭进程高度相关的候选基因。
体外CRISPR筛选技术:
构建针对候选基因的慢病毒文库,在原代CD8+ T细胞中进行功能缺失筛选,结合功能性读数(如细胞因子分泌、抑制性受体表达),鉴定对T细胞耗竭具有调控作用的关键基因。
分子生物学与生物化学:
通过免疫共沉淀(Co-IP)结合质谱分析,鉴定KLHL6的底物蛋白;利用泛素化实验验证KLHL6对底物的降解作用。
细胞代谢分析:
通过Seahorse能量代谢分析仪和共聚焦显微镜,评估线粒体的呼吸功能及动态变化(裂变/融合)。
动物模型验证:
利用过继性T细胞转移模型(如肿瘤模型和LCMV慢性感染模型),验证调控KLHL6对T细胞抗肿瘤/抗病毒功能及耗竭状态的影响。
研究路线
差异基因筛选:
利用已有T细胞转录谱表征并区分急性感染(效应/记忆分化)与慢性感染(耗竭分化)下的差异基因,锁定E3连接酶相关基因介导的蛋白质稳态在T细胞耗竭与线粒体维持中的关键作用。
CRISPR文库筛选与验证:
通过体内CRISPR文库筛选与体外敲除/过表达双重验证,确认KLHL6是T细胞耗尽和线粒体功能的关键调控因子。
底物筛选与机制解析:
寻找并验证KLHL6直接作用的底物蛋白,发现其靶向降解TOX(T细胞耗竭的关键转录因子)并调控PGAM5-Drp1线粒体裂变轴。
代谢与功能关联分析:
明确KLHL6缺失导致的线粒体功能障碍如何进一步加剧T细胞功能衰退。
临床转化意义:
在肿瘤模型和慢性感染模型中,评估增强KLHL6表达对免疫治疗效果的提升作用。
主要结果
1.KLHL6是T细胞耗竭与线粒体功能障碍的双负调控因子
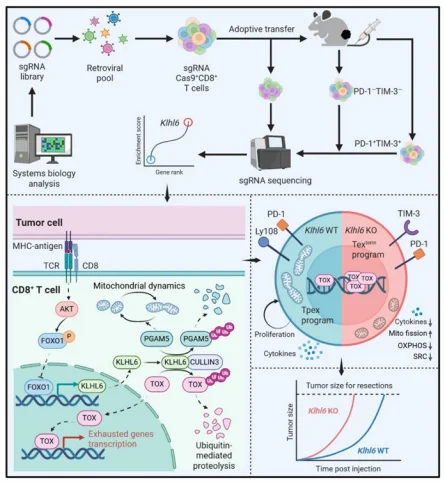
作者将八项独立感染和癌症研究的136份CD8+T细胞的整体RNA测序(RNA-seq)样本中数千个基因的转录变异提炼为两个核心差异基因模块,以定义慢性(肿瘤和/或感染)与急性(感染)条件下CD8+T细胞的全转录组特征,并确认了E3连接酶相关基因介导的蛋白质稳态在T细胞耗竭与线粒体维持中的重要作用。针对78个与T细胞耗竭负相关,以及133个与线粒体功能正相关的E3连接酶相关基因的两项平行体内CRISPR文库筛选共同确认KLHL6是T细胞耗尽和线粒体功能障碍的双负调控因子。
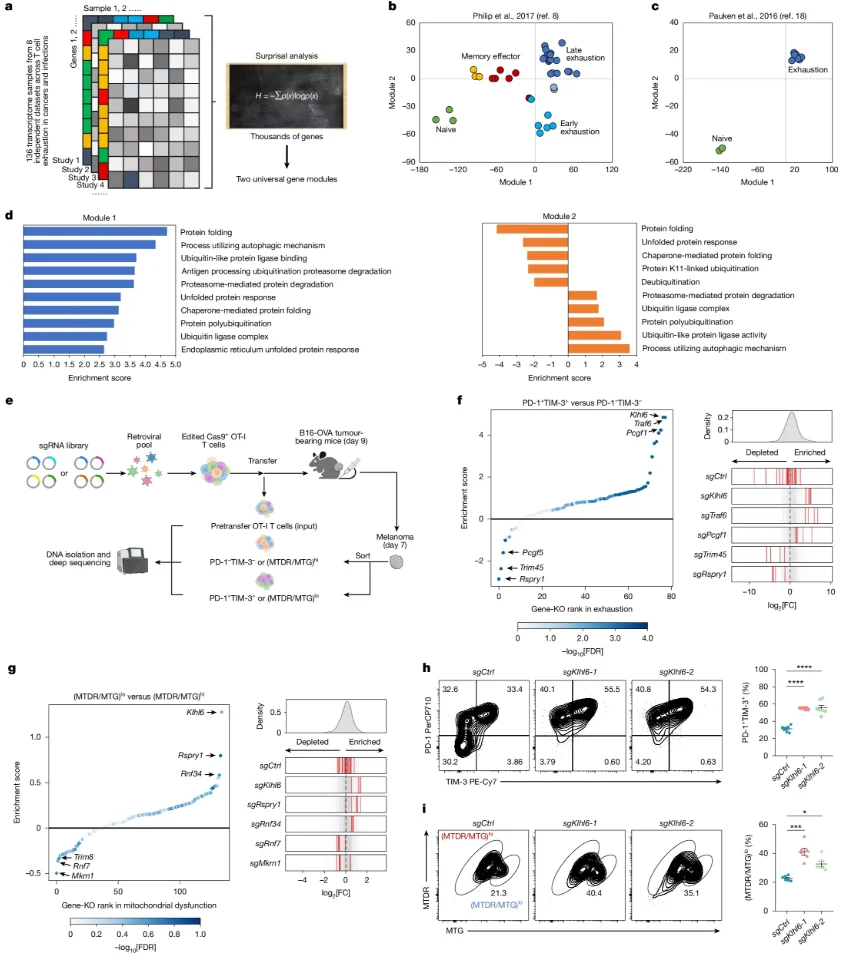
图1. KLHL6是T细胞耗竭与线粒体功能障碍的双负调控因子
2.KLHL6缺乏通过TOX蛋白累积来驱动T细胞终末耗竭分化
建立Klhl6−/− OT-I小鼠进一步表征KLHL6缺乏对T细胞功能的影响发现,KLHL6缺乏导致肿瘤、引流淋巴结和脾脏中CD8+T细胞积聚减少、高表达PD-1和TIM-3的终末耗竭T细胞增多,以及肿瘤坏死因子(TNF)和IFNγ的分泌变少。
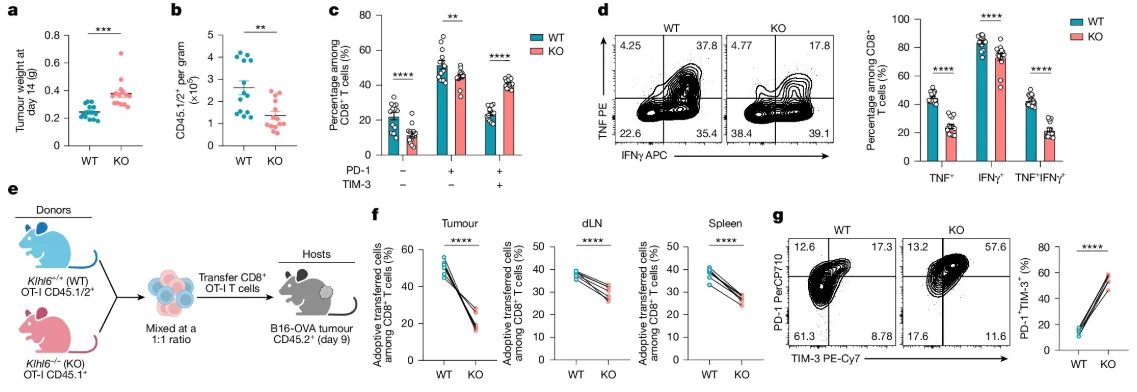
图2.KLHL6缺乏促进T细胞耗尽并损害其功能
相反,接受KLHL6-OE OT-I T细胞过继治疗的B16-0VA小鼠中肿瘤、引流淋巴结和脾脏中CD8+T细胞积累增加、终末耗竭T细胞的比例降低、TNF和IFNγ的分泌变多,以及生存期延长。这些数据共同表明KLHL6的丧失导致CD8 +T细胞趋向衰竭和功能障碍。
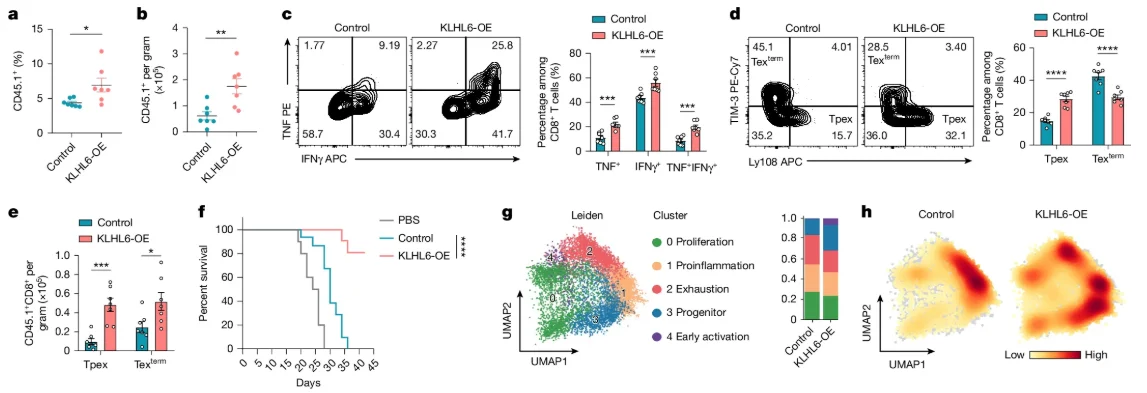
图3. KLHL6抑制Tpex细胞向终末分化的转变,增强抗肿瘤免疫
3. TOX作为KLHL6的下游靶点,促进Tex细胞的末端分化
进一步的机制研究发现,TOX作为KLHL6的下游底物在T细胞耗竭中显著富集。KLHL6缺乏或过度表达通过泛素化显著调控了受刺激和未受刺激T细胞中的TOX蛋白水平。既往研究已表明TOX是启动和促进T细胞耗竭的关键调控因子。综合316名不同癌症类型的患者TILs的泛癌scRNA-seq数据发现KLHL6介导的TOX蛋白水平调控了前体耗竭T细胞向终末耗竭T细胞的转化,进一步支持KLHL6作为T细胞抗肿瘤功能关键调控因子的机制作用。
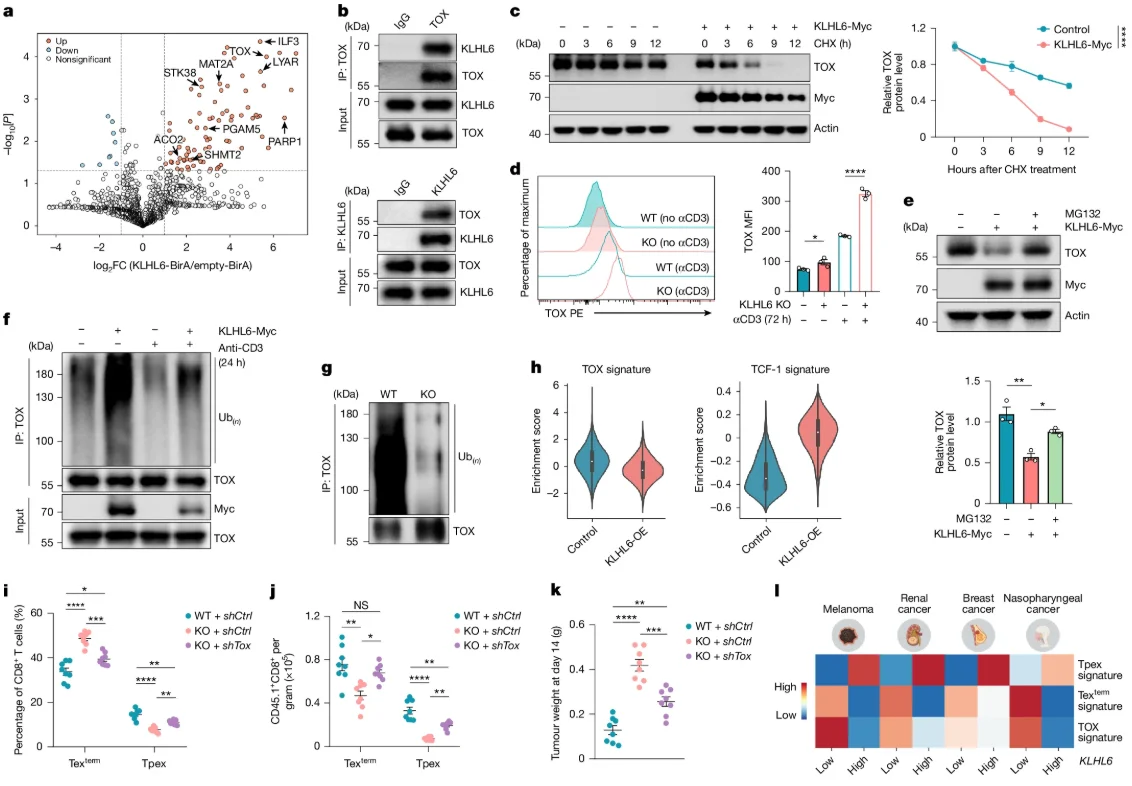
图4. TOX作为KLHL6的下游靶点,促进Tex细胞的末端分化
4.KLHL6通过调控PGAM5-Drp1信号轴来限制线粒体过度分裂对T细胞的功能损害
KLHL6缺失的T细胞比WT细胞具有更小且嵴短的线粒体,反映了线粒体裂变与融合之间的平衡向过度裂变转变。在KLHL6下游底物中,磷酸甘油酸突变酶5(PGAM5)的敲降恢复了KLHL6-KO T细胞中融合相关蛋白(Mfn2、Opa1)和磷酸化Drp1表达水平,改善了线粒体活性。
综上,KLHL6主要通过限制TOX驱动的耗尽分化和PGAM5介导的线粒体功能障碍,支持T细胞抗肿瘤功能。仅用TOX或PGAM5的KD部分恢复抗肿瘤免疫,改善肿瘤进展,增强TIL积累,减少终末耗竭并增加细胞因子生成。而双重KD使得几乎完全的救援,表型和功能特征基本恢复到WT水平。这些发现进一步证实TOX和PGAM5是KLHL6调控T细胞耗竭和抗肿瘤免疫的关键功能底物。
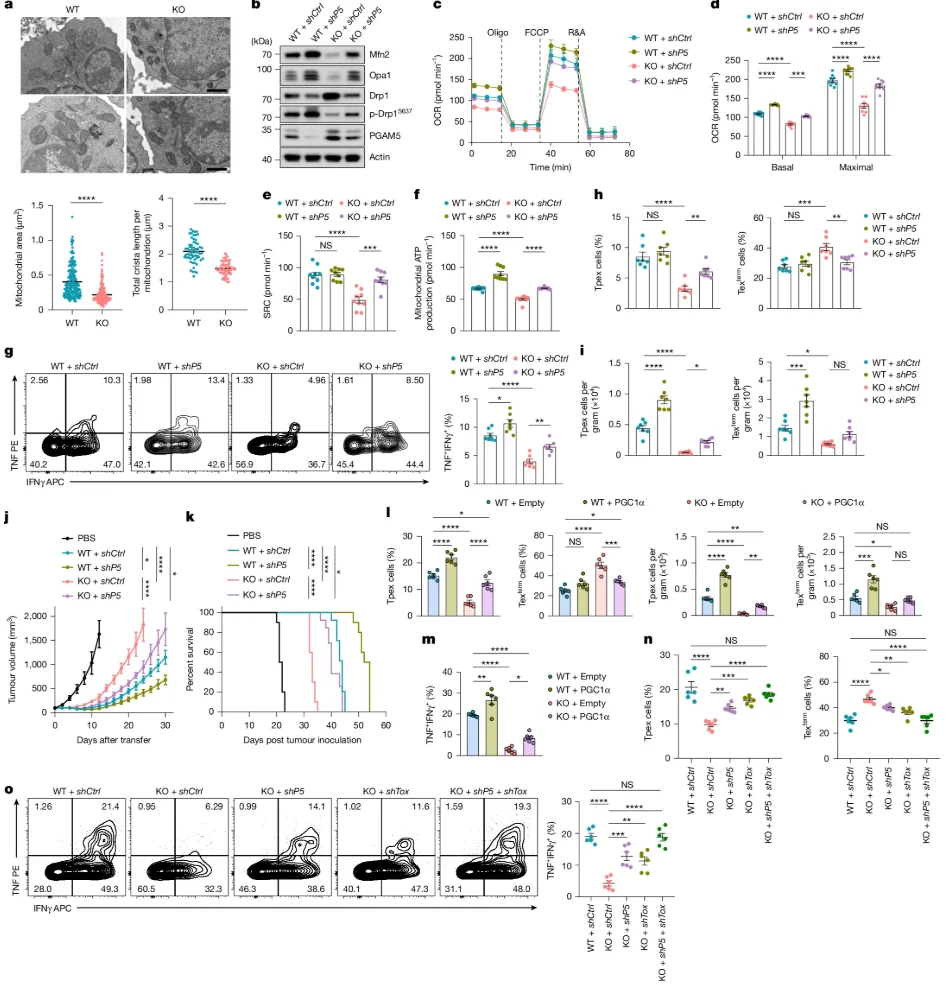
图5. KLHL6通过PGAM5调控T细胞线粒体稳态来调节抗肿瘤反应
研究意义与创新点
- 理论创新: 首次将E3泛素连接酶KLHL6确立为连接持续性TCR信号、转录重编程(TOX降解)与代谢重塑(线粒体动力学)的关键枢纽,解答了“持续性信号如何驱动耗竭”这一基本问题。
- 靶点价值: 揭示了KLHL6作为一个“多功能”靶点的潜力。不同于以往单一靶向抑制性受体的策略,增强KLHL6可以同时纠正T细胞的转录耗竭程序和代谢缺陷。
- 治疗启示: 为免疫治疗提供了新的思路,即通过稳定或增强KLHL6的表达,或者TOX/PGAM5双重抑制,有望突破CAR-T和TCR-T疗法面临的耗竭瓶颈,开发出更持久、更有效的下一代细胞疗法。
文章小结
本研究通过整合CRISPR筛选与机制解析,鉴定出E3泛素连接酶KLHL6是CD8+ T细胞抵抗耗竭的关键“守门人”。 KLHL6通过双重机制——靶向降解耗竭驱动因子TOX以及调控线粒体稳态——来维持T细胞的持久战斗力。 该发现不仅从蛋白质稳态调控层面深化了对T细胞耗竭机制的理解,更为重要的是,它提出了一个极具转化潜力的治疗靶点,为改善肿瘤免疫疗法的疗效开辟了新路径。
源井生物CRISPR-iScreen™文库
CRISPR-iScreen™是源井生物自主研发的一项创新技术,旨在实现高效的CRISPR筛选。目前,源井生物拥有超过40种的CRISPR文库现货供应,还提供一站式CRISPR文库(体内/体外)功能筛选服务,随时支持您的研究需求。
联系我们了解更多详情 >>>






